Abstract
An excipient’s functional category is a qualitative classification or term that describes the purpose or role of an excipient in a drug product. However, the current regulatory environment and the paradigm of QbD go beyond simply identifying excipient function and emphasizes performance through the identification, evaluation, and control of critical material attributes (CMA) that assure consistent performance throughout a product’s life-cycle [1]. Not all performance-related CMA of an excipient may be identified or evaluated by specific tests and specifications listed in compendial monographs [2]. With this in mind, USP Excipient Performance chapter <1059> [3] is designed to provide an overview of typical material attributes for many functional categories along with additional tests that may be useful in evaluating and controlling excipient attributes that are not typically included in compendial monographs. Selection of the appropriate tests and specifications that are necessary to ensure consistent and reliable excipient performance requires an understanding of the formulation and manufacturing processes, the dosage form performance requirements, and the physical and chemical properties of each ingredient in the dosage form.
Introduction
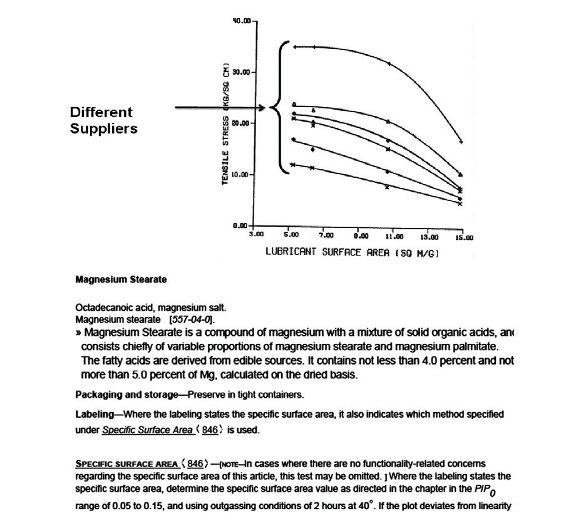
Figure 1 - Why Excipient Performance Testing? A classic Example (Magnesium Stearate) [4,5] Surface area is well-known to influence the lubrication properties of magnesium stearate and this can impact product properties – in this case tablet tensile strength
Setting compendial standards for excipients can oftentimes be more challenging than setting standards for active pharmaceutical ingredients (APIs). Unlike APIs, excipients may not be specifically made for use in specific drug products, are often available from multiple suppliers, and are often manufactured for multiple industries other than the pharmaceutical industry such as the food, cosmetic, or personal care industries. In addition, the quantity of ingredients sold for pharmaceutical applications compared to total sales may be sufficiently small that the cost of setting and maintaining appropriate compendial standards becomes cost prohibitive. Other challenges can arise because excipients that are manufactured specifically for the pharmaceutical product industry sometimes have special grades available (e.g., microcrystalline cellulose, magnesium stearate (Figure1) [4,5]) that do not have monograph specifications (e.g., particle size, surface area). Yet another challenge is multisource suppliers of the same grade of excipient which can lead to batch-to-batch or supplier-supplier variability and, potentially, inequivalent performance. The successful manufacture of a robust product requires the use of well-defined excipients and processes that together yield a consistent product [6]. Finally, a vast array of excipient applications exists in pharmaceutical product development and an excipient may have any of a number of purposes depending on its use in a formulation, manufacturing process, and dosage form. As a result, selection of appropriate performance tests requires a sound understanding of the role of the excipient and the CMA relevant to a dosage form. The requirements for one material and formulation may not be suitable for the same material in another formulation.
Table 1 - Some Important Physical, Chemical and Mechanical Properties Affecting Excipient Function and Performance:

USP and NF monographs for excipients, indeed for most pharmacopeial monographs, are primarily concerned with chemical identification, purity, and quality with few requirements for physical or chemical properties that relate specifically to excipient function or performance. Industry has long recognized this challenge and knowledge gap. Yet, the development, manufacture, and performance of pharmaceutical dosage forms depend heavily the excipients physical and chemical properties. Excipients that are manufactured and supplied to comply with compendial standards may, therefore, not have all the CMA identified or evaluated in monographs using compendial test methods, procedures, or acceptance criteria (monograph specifications) (Table 1). Excipient suppliers and users must therefore independently identify and control excipient CMAs that go beyond monograph specifications. Selection of tests and appropriate specifications that are necessary to ensure consistent and reliable excipient performance requires a thorough understanding of the physical and chemical properties of the excipient, formulation, and manufacturing process. Excipient users should anticipate lot-to-lot and supplier-to-supplier variability in excipient properties and therefore should have appropriate controls in place to ensure consistent excipient performance.
The above terms are defined here to clearly understand the differences [7]:
- Excipient functional category: the general purpose or utility of an excipient (function) in a formulation (e.g., diluent, lubricant, glidant).
- Excipient performance: determined by the physical, chemical or mechanical properties that influence an excipient’s ability to perform its intended function in a formulation.
- Excipient performance test methods: quantitative physical, chemical, or mechanical test methods that assess properties of an excipient that influence its ability to perform the intended function in a formulation.
- Excipient functionality: sometimes used to describe excipient functional category and sometimes used to describe more quantitative performance-related attributes. Because of the ambiguous nature of the definition, use of the term ‘excipient functionality’ is not encouraged.
Traditional vs. QbD Approach to Product Development
Traditionally, the pharmaceutical industry focused on the use of similar excipient lots, for example, a single supplier and a specific grade, during development and manufacturing in an effort to minimize variability. This practice often led to an “optimized” formulation that met compendial specifications and product performance requirements but locked in or created a “frozen” process. Despite the general awareness of the importance of CMA for excipients in controlling variability of the final product attributes, this practice did not provide a knowledge base of the role played by many of the CMAs of pharmaceutical excipients that relate to drug product critical quality attributes (CQA). As a result, there was limited progress in creating standard methods for characterizing pharmaceutical excipients [8].
In 2002, FDA launched a new initiative “Pharmaceutical CGMPs for the 21st Century” [9] to integrate quality systems and risk management approaches into its existing programs with the goal of encouraging industry to adopt modern and innovative manufacturing technologies [10]. In 2006, FDA issued a guidance to help manufacturers implement modern quality systems and risk management approaches to meet the requirements of the Agency’s CGMP regulations [11]. The guidance elaborated key concepts integral to the quality systems approach that includes Quality-by-Design (QbD) and Product Development [12], quality risk management [13], corrective action and preventive action (CAPA), change control, and the six-sigma inspection model. ICH Q8 and these regulatory initiatives have created a regulatory environment that is supportive of the pharmaceutical industry’s developing and sharing with the regulatory agencies, an enhanced knowledge of product performance over a wider range of material attributes, processing options, and process parameters. QbD [8] emphasizes the development of an understanding of the role and impact of excipient properties on product CQA and applications of these approaches are now appearing in the scientific literature taking advantage of risk analysis, experimental design, and historical data to assess the impact of excipient attributes on critical product attributes [14,16].
Understanding material properties and processing can build a robustness and flexibility into the manufacturing process that, together with appropriate specifications, better ensures product quality. The aim of pharmaceutical development within the QbD paradigm is to design a quality product from the ground up by understanding and controlling excipient CMAs and to integrate this into the product manufacturing process in a way that will consistently deliver the intended product performance.
20 Years of Functionality, Physical Testing, and Excipient Performance
Since the 1970’s, USP Excipient Expert Committees have struggled with the question of whether or not “functionality” should be included in excipient monographs and, if so, which ones and how [17]. In an effort to address these questions, the USP Excipients Expert Committee established an advisory panel on Physical Test Methods for excipients in 1991 to address “functionality.” The advisory panel, recognizing that excipient monographs focused on strength, quality, purity and identity but not excipient performance focused its efforts on quantitative, performance-related tests that were, in effect, physical tests. The issue was not so much related to functionality or functional category, since lots from two different suppliers of USP Magnesium Stearate would likely function as a lubricant but rather, one of consistency of performance. In 2000, the Excipients General Test Methods Expert Committee was formed and continued the work by adding or updating 20+ test methods and information chapters and modified several excipient monographs through labeling requirements. It has also harmonized excipient-related general chapters with both the European Pharmacopoeia and Japanese Pharmacopoeia through the Pharmacopieal Discussion Group (PDG) [18].
USP introduced a survey at the 2005 USP Annual Science Meeting (ASM) [19] to excipient users and manufacturers to assess the need for performance testing. Results of the survey were presented along with a joint discussion on USP and European Pharmacopoeia perspectives at the 2006 ASM meeting [20]. Also in 2005, the USP Excipients General Chapters Expert Committee published a Stimuli article [21] on the development of a General Information Chapter, <1059> Excipient Performance. As a result, a Joint Advisory Panel was established by the USP Excipient Collaborative Group (Excipient Monograph 1, Excipient Monograph 2 , and Excipient General Chapter Expert Committee’s) charged with the development of an information chapter on Excipient Performance.
Following extensive work by the Advisory Panel, the publication of a proposed Information Chapter <1059> Excipient Performance [22] was announced at the 2009 USP International Excipient Workshop [23]. The general information chapter became official in USP 33 – NF 28, 2nd Supplement, February 2011. In the current revision cycle, the Physical Analysis Expert Committee continues the review and revision of <1059> through an Expert Panel (EP).
In parallel with USP efforts, the European Pharmacopoeia (Ph. Eur.) published chapter 5.15. Functionality-Related Characteristics of Excipients [24] in 2005 and introduced a non-mandatory section, Functionality-related Characteristics (FRC’s) to approximately 100 excipient monographs to control CMA. This chapter and the FRC sections in specific monographs are not mandatory and are published for information and guidance. Thus, Ph. Eur. provides guidance on critical material attributes using a different compendial approach.
Chapter <1059> Excipient Performance
Table 2 - NF excipient categories included in <1059> Excipient Performance

Chapter <1059> Excipient Performance provides an overview of the key functional categories of excipients in USP-NF along with test methods and procedures that may be used to assess excipient function and performance. Each functional category includes a general description; the mechanisms by which the excipients achieve their intended function; physical properties common to these excipients; chemical properties; and a list of pharmacopeial general chapters that may be useful in the development of specific tests, procedures, and acceptance criteria. A list of excipients is also available in USP-NF grouped by functional categories under Contents [25]. The list of excipients is not comprehensive, however, and is not intended to limit the choice or use of any material. The functional categories included in <1059> (Table 2) are organized by their most typical use in common pharmaceutical dosage forms (Tablets and Capsules; Oral Liquids; Semisolids, Topicals and Suppositories; Parenterals; and Aerosols) to provide a greater level of focus for each functional category. Some functional categories can, of course, apply to multiple dosage form types. The association of a functional category with a particular dosage form or excipient is not absolute and is not intended to limit use. Because of the complex nature and interactions of dosage form ingredients, processing, and dosage form performance requirements, the information provided in this chapter should be viewed as a general guidance and is neither restrictive nor completely comprehensive. Figure 2 provides the general layout of each dosage form and associated functional category listing.

Figure 2 - <1059> Excipient Performance Functional Category Format
<1059> Chapter is intended to help formulation scientists identify and ‘monitor’ properties that they determine are important to excipient function and performance. It also provides a common methodology that can facilitate communication between the supplier, user and regulatory agencies. The information on excipient performance can be used, as appropriate, to justify the choice and quality attributes of the excipient and to support the justification of the drug product specification.
Future Revision of <1059> Excipient Performance
The <1059> Excipient Performance Expert Panel is currently working on inclusion of additional dosage forms and NF categories. The proposed revision to <1059> is scheduled to appear in 2012 [PF 38(3)] with a plan to include an additional four dosage forms and thirteen NF functional categories. The additional dosage forms and functional categories are:
- Dosage forms: Radiopharmaceuticals, Ophthalmic Preparations, Dry Powder Inhalers, Transdermals and Patches.
- NF Categories: Adhesive, Bulking Agent for Freeze-drying, Carrier (Vehicle), Colloidal Stabilizing Agent, Film-forming Agent, Flavors and Perfumes, Free-radical Scavengers, Humectant, Penetration Enhancer, Reducing Agent, Releasemodifying Agent (hydrophilic; hydrophobic), Transfer Ligand, and Water-repelling Agent.
The <1059> Excipient Performance Expert Panel will also propose a revision to the USP reference table “Excipients - USP and NF Categories, listed by category” in the National Formulary to align with the <1059> NF category proposed changes. The purpose of updating the USP-NF category listing of excipients is to reflect the general chapter’s approach of summarizing the functional capabilities of an excipient under their appropriate dosage form in order to provide the reader a general overview of each NF functional category. The table will be an extension of this general chapter to aid the reader in listing the excipients under a dosage form and their most typical function.
Summary
Excipients are critical to the performance of the dosage form yet not all excipients are the same. Furthermore, the same excipient does not always perform the same function [26].
Chapter <1059> provides an overview of the key functional categories of excipients identified in USP–NF and provides guidance about the most common properties that might be important for a particular material in a particular application. The chapter provides reference to standard USP test procedures that can be used by both the manufacturers and users. The non-mandatory nature of <1059> avoids possible confusion with mandatory tests and labeling tests that are the subject of specific USP excipient monographs. The chapter does not impose limits or specifications since the properties of an excipient that are required will vary and depend upon the product, manufacturing process, quantity, and intended function.
In summary, the USP-NF Chapter <1059> on Excipient Performance is intended to help users by:
- providing guidance on properties that may be important for a particular material in a particular application.
- providing reference to standard USP methods that can be used by both manufacturers and users to facilitate communication while keeping tests non-mandatory. avoiding confusion with mandatory tests and labeling tests.
- not imposing limits/specifications.
References
- FDA Guidance for Industry Q8 (R2) Pharmaceutical Development http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073507.pdf Accessed September 15, 2011.
- USP 34 – NF 29 (United States Pharmacopeial Convention) General Notices, 4.10 Monographs. USP, Rockville, MD, USA, 2009, p. 4.
- USP 34 – NF 29 (United States Pharmacopeial Convention) General Information Chapter, Excipient Performance <1059>, Rockville, MD: USP; 2011, pp. 555-565.
- Dansereau, R.; Peck, G.E. The effect of the variability in the physical and chemical properties of magnesium stearate on the properties of compressed tablets. Drug Dev. Ind. Pharm. 1987, 13 (6), 975–999.
- Magnesium stearate NF, USP 34 – NF 29, (United States Pharmacopeial Convention). USP, Rockville, MD, USA. 2009, pp. 1571-157.
- USP 34 – NF 29 (United States Pharmacopeial Convention) General Information Chapter, Good Manufacturing Practices for Bulk Pharmaceutical Excipients <1078>, Rockville, MD: USP; 2011.pp 583-595.
- Sheehan, C. USP Excipient Performance Chapter <1059>: Excipient QbD as It Relates to Performance and Functionality. ExcipientFest 2011, Baltimore MD.
- L.X. Yu. Pharmaceutical Quality by Design: Product and Process Development, Understanding, and Control. Pharmaceutical Research. 25:781-791 (2008).
- Pharmaceutical Quality for the 21st Century A Risk-Based Approach Progress Report Department of Health and Human Services U.S. Food and Drug Administration May 2007 http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm128080.htm Accessed September 15, 2011 .
- Pharmaceutical CGMPS For The 21st Century — A Risk-Based Approach - Final Report.http://www.fda.gov/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswersonCurrentGoodManufacturingPracticescGMPforDrugs/ucm137175.htm Accessed September 15, 2011.
- FDA Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulations http://www.fda.gov/downloads/Drug s/GuidanceComplianceRegulatoryInformation/Guidances/ucm070337.pdf Accessed September 15, 2011.
- FDA Guidance for Industry Q9 Quality Risk Management http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm128053.pdf Accessed September 15, 2011.
- FDA Guidance for Industry Q10 Pharmaceutical Quality System http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073517.pdf Accessed September 15, 2011.
- F. Yacoub, J. Lautens, L. Lucisano, and W. Banh. Application of Quality by Design Principles to Legacy Drug Products. Journal of Pharmaceutical Innovation:6:61-68 (2011).
- A.J. Hlinak, K. Kuriyan, K.R. Morris, G.V. Reklaitis, and P.K. Basu. Understanding critical material properties for solid dosage form design. Journal of Pharmaceutical Innovation: 12-17 (2006).
- J. Kushner, B.A. Langdon, J.I. Hiller, and G.T. Carlson. Examining the impact of excipient material property variation on drug product quality attributes: a quality-by-design study for a roller compacted, immediate release tablet. Journal of Pharmaceutical Sciences. 100:2222-2239 (2011).
- Shangraw, R. F. Compendial Standards For Pharmaceutical Excipients. Drug Development And Industrial Pharmacy. Vol. 13(13), 2421-2439. 1957.
- Pharmacopeial Discussion Group (PDG). USP International Pharmacopeial Harmonization http://www.usp.org/USPNF/pharmacopeialHarmonization/ Accessed September 15, 2011.
- Amidon, G. E. Excipient Performance Tests. USP Annual Science Meeting, San Diego, CA. 2005.
- Amidon, G.E. Performance Related tests in Excipients. USP Annual Science Meeting, Denver, CO. 2006.
- USP Pharmacopeial Forum: 2007; PF 33(6) [Nov.-Dec. 2007] p.1045. (United States Pharmacopeial Convention). PF 33(6) Stimuli to the Revision Process: Proposed New USP General Information Chapter, Excipient Performance <1059>.
- USP Pharmacopeial Forum: 2009. (United States Pharmacopeial Convention). PF 35(5) [Sept. –Oct. 2009] p. 1228. Excipient Performance <1059>.
- Amidon, G.E. The Future of USP Performance Testing at USP: Excipient Performance Chapter <1059>. 2009. USP International Excipient Workshop, Excipient Quality Control, Testing, and International Harmonization. USP Headquarters, Rockville, MD.
- European Pharmacopoeia, Seventh edition (7.2) 2011. 5.15. Functionality-Related Characteristics of Excipients. Accessed September 15, 2011.
- USP 34 – NF 29 (United States Pharmacopeial Convention). USP and NF Excipients, Listed by Category. Rockville, MD: USP; 2011, pp. 1415-1420.
- Amidon, G.E. Excipient Performance <1059>, Critical Material Attributes, QbD and Functionality Related Characteristics. USP Sponsored Workshop at 2010 AAPS-PSWC Meeting. Impurities, Adulteration, and the Changing Role of the USP in Global Drug Quality, November, 2010. New Orleans, LA.
Author Biographies
Dr. Amidon received his Bachelor of Science degree in Medicinal Chemistry (1974) and his Ph.D. in Pharmaceutical Chemistry (1979) from the University of Michigan at Ann Arbor, MI. He is currently Research Professor of Pharmaceutical Sciences at the University of Michigan, College of Pharmacy in Ann Arbor. Dr. Amidon has over twenty-eight years of industrial research and development experience with Pfizer, Inc., Pharmacia, Pharmacia & Upjohn, and originally The Upjohn Company in Kalamazoo, MI. He is recognized for his expertise in the physical, chemical and mechanical property characterization of active pharmaceutical ingredients, excipients and products, as well as the development of scientific strategies for oral solid dosage form development. Dr. Amidon has been an active member of the American Association of Pharmaceutical Scientists and USP throughout his career. He has served USP since 1990 in various capacities and is currently Chair of the Physical Analysis Expert Committee. Dr. Amidon is a member and Fellow of the American Association of Pharmaceutical Scientists and a past recipient of the Ebert Prize from the American Pharmaceutical Association.
Ms. Sheehan received her B.Sc. degree from the National University of Ireland, University College Cork and her M.S. in Bioscience Regulatory Affairs from Johns Hopkins University, Baltimore, MD, USA. She is currently the Director of Excipients and in her current role, is responsible for the development and update of excipient monographs and related chapters and participates in the Pharmacopeial Discussion Group’s compendial harmonization of excipient monographs and related chapters. She also shared responsibility for the successful launch under USP of the Food Chemicals Codex (FCC) Sixth Edition on February 29, 2008. Ms. Sheehan has over 15 years of experience in organic chemistry, biochemical, microbiological and biotechnology, industry related testing. Ms. Sheehan has been an active member in AAPS, PDA, CHPA, and RAPS. She was a working member on the Product Quality Research Institute Excipient Working (PQRI) Group from 2006-2007. The Group published two articles entitled, “PQRI Survey of Pharmaceutical Excipient Testing and Control Strategies Used by Excipient Manufacturers, Excipient Distributors and Drug Product Manufacturers”, September 2006 and, “PQRI Joint Position Paper on Pharmaceutical Excipient testing and Control strategies”, September 2007.
This article was printed in the September/October 2011 issue of American Pharmaceutical Review - Volume 14, Issue 6. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.