By: Kim Huynh-Ba* - Managing Director - Pharmalytik, LLC and Chris Latoz - Stability Manager - Hollister Incorporated
*corresponding author
Abstract
The purpose of a Stability Program is to support the expiration dating of pharmaceutical products, medical devices, and biologics and recommend storage conditions. These products are manufactured and placed in special environmental chambers (stability chambers) with specific temperature and humidity conditions according to the ICH Q1A (R2) guideline. Samples are then withdrawn and tested at various time intervals, according to specifications. Stability data are generated throughout the product lifecycle to support development, commercialization, distribution, and even transportation. The stability data sets are part of regulatory submissions; thus, any deviation or investigation may cause sample integrity concerns. According to the FDA, WHO, and various global guidelines, the temperature and humidity of the stability chambers must be accurately controlled to specific tolerances. Excursions must be evaluated and investigated if they exceed 24 hours in duration and if there is any impact on the quality of the products, additional actions may be necessary. This paper will provide some key factors to consider regarding how temperature and humidity excursions to stability samples should be investigated and what data is needed to support conclusions about sample viability. This article may also be of interest to the chamber management group of the Stability Program and those involved with facility management. Various suggestions can be incorporated into a company’s internal standard operating procedure (SOP) to handle deviations and protect the integrity of products stored.
Introduction
ICH Q1A (R2) provides the minimum requirements of stability data packages for registering new drug substances and drug products. Stability studies are conducted using various temperature and humidity-controlled chambers to provide data needed for establishing a commercial expiry of the drug product, or biologic. Although the ICH document does not include medical devices in its scope, many principles included can also be expanded into medical device stability studies. Representative stability samples are stored in these conditions and withdrawn according to a pre-approved testing plan or protocol. Testing is performed on these samples using validated analytical procedures, and data is evaluated against relevant product specifications to support the expiration dating of pharmaceutical products. According to the guidance from the Food and Drug Administration (FDA), the World Health Organization (WHO), the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH), and various global health authorities, these storage conditions require that the temperature and humidity of the stability chambers must be controlled within ± 2°C and ±5% Relative Humidity (RH). Excursions outside of these variations must be addressed and investigated.1,2,3 For injectables or biologics that are kept frozen, the temperature may need to be controlled within ± 5°C or ± 10°C, with no humidity requirement.
Not addressing chamber excursions is a frequent audit finding, as is evidenced by a sampling of Warning Letters from the FDA, including:
“Chart recorders indicate that stability chamber temperatures also fluctuated wildly without any explanation or investigation on multiple occasions for months at a time. Chart recorders also indicated that the stability chambers’ actual temperatures were different from their intended temperatures on multiple studies for extended periods.”4
“Additionally, your firm failed to investigate excursions of temperature and relative humidity in your stability chambers and sample retention room and failed to investigate incorrect labeling of laboratory samples that led to laboratory errors.”5
“No follow-up or investigations were conducted for the excursions listed above to determine root cause and potential impacts on the products and stability studies.”6
“The inspection documented that, despite the fact that your firm has an uninterrupted power supply used by the QC laboratories, power failures have impacted the QC stability chambers. However, in each case, no investigation was conducted to determine the impact of the power loss on the samples kept within the chambers.”7
Since the testing ultimately establishes the commercial expiration dating of the products and is part of global regulatory submissions, any data must be accessible during any audit or inspection. Therefore, any deviation of the storage chambers will impact the quality systems and potentially multiple product lines. A risk management program should be used with additional tools including:
- Applying Mean Kinetic Temperature (MKT)
- Using data from stress studies
- Adjusting the stability sample pull schedules
- Testing extra product samples for additional data
While plenty of information addressing temperature excursions during distribution and transportation exists, there is limited literature on addressing stability chamber temperature excursions. A recent presentation by Anderson and Seevers on July 9, 2020, discussed how the United States Pharmacopeia Convention (USP) uses Mean Kinetic Temperature (MKT) to evaluate and address temperature and humidity excursions in the storage of pharmaceutical products.8 However, there is a lack of general guidance on investigating stability chamber excursions and determining the impact on product samples. Many questions remain, such as:
- How many excursions are acceptable through the length of the stability study?
- Are 24 hours the length of each excursion or the cumulative total time allowed to be outside of the controlled range?
- How will the deviation be addressed?
- How can an investigation plan be determined?
- What data can be used to support an investigation of chamber Excursions?
Regardless of how well stability chambers are constructed and maintained, inevitable deviations, including power failures, chamber component failures, even human error (i.e., a technician fails to completely close the chamber door) will eventually happen. The result is a temperature and/or humidity excursion, and the deviation must be addressed to determine the impact.
Consider the scenario where a technician leaves a chamber door open on an afternoon; the excursion can easily extend beyond 24 hours, triggering an investigation. A very important initial step to conducting the investigation is knowing precisely what samples are stored in the chamber. Thus, keeping accurate, up-to-date chamber sample inventories is of paramount importance.
Once an excursion has occurred, it is critical to know the total cumulative time out-of-range and the worst case cold or warm temperature, depending on the directionality of the excursion (according to the environmental monitoring system) as well as its length. It is also critical to know the current inventory, especially the temperature-sensitive products. As an example, a weekend excursion may not be critical for a 12-month sample, however, it may have a greater impact on a 1-month sample.
It is also important to consider the impact on humidity if any samples are in permeable packaging, as relative humidity is a function of temperature. Knowing these three details (duration, worst case temperature, presence of permeable packaging) will help drive the investigation, the corrective action, and the immediate remedial action if needed. Stability characteristics of the products are also critical in the evaluation of these excursions; therefore, labile products may be exposed to a higher impact than stable products and should be considered on a case-by-case basis.
Establish a Risk Assessment Evaluation
It is critical to understand the inherent physical and chemical characteristics of the impacted sample material. This knowledge will be the foundation to assess the risks and impact of the excursions to the stability program and allow for taking and prioritizing appropriate actions. Figure 1 lists essential information that should be available to establish a risk assessment and evaluate the impact of any excursion. Many risk management tools can be used to identify the critical attributes that impact product stability after excursions occur.16

Special attention should be made to temperature/moisture-sensitive products. Biologics generally cannot tolerate excess excursions as they may lose their therapeutic properties completely. Repeating a stability study due to an excursion is not a desirable option due to the loss of time and product, and the risk of generating conflicting data. Additionally, some degradation processes of biologics are only detected at higher stress temperatures, making it irrelevant to the shelf-life determination to be used for the temperature excursion evaluation. The appropriate controls and due diligence offer a scientific and technically valid approach to disposition the continued integrity of any ongoing study.
Stability Chamber Excursion Scenarios and Recommendations
Table 1 lists several recommendations depending on the nature of the excursion that occurred, and the chamber (Controlled Real Time (CRT) or Accelerated) affected. It is noted that refrigerated and frozen conditions are not addressed in this paper. In many cases, Mean Kinetic Temperature (MKT) can describe the chamber performance in a longer period (30 days or more). These data would apply to all the products stored in these chambers. Since different products may have different degradation profiles, an immediate risk assessment will help to identify the most vulnerable studies which may be protected by chamber transfer and/or immediate testing to verify the baseline impact of the excursion.

As indicated in Table 1, four options are recommended depending on the excursions, data available, and the stability of the samples stored. It is recommended that justification should be documented as part of the risk assessment of the excursion as well as evidence for the corrective and preventive actions (CAPA) taken.
Using Mean Kinetic Temperature (MKT)
When the temperature of a CRT chamber falls outside of tolerance for a period of more than 24 hours, the concept of Mean Kinetic Temperature (MKT) may help in determining the impact of the stability chamber excursion on product samples. Generally, you would want to use MKT for CRT stability chambers only, as the kinetics of product degradation may change, and other routes of degradation may become possible at accelerated or stress temperatures.9 It is especially important to consider the impact of approaching a phase change (melting point, deformation point, glass transition temperature, etc.) for the stored sample materials. Most stability programs strive for a +10°C buffer range for known phase changes.
MKT, as defined in ICH Q1A(R2), is a single derived temperature that, if maintained over a defined time period, affords the same thermal challenge to a drug substance or drug product as would be experienced over a range of both higher and lower temperatures for an equivalent defined period.2 Described slightly differently in USP<1079> Good Storage and Distribution Practices for Drug Products, “The MKT is the single calculated temperature at which the total amount of degradation over a particular period is equal to the sum of the individual degradations that would occur at various temperatures.” In general terms, the MKT converts a variable temperature into an equivalent steady temperature. Then, the steady temperature is used to determine the effect of the excursion on product quality.10
J.D. Haynes is credited with first developing the formula for calculating MKT,11 which will always be higher than the arithmetic means temperature. The equation applies the Arrhenius equation and the logarithmic temperature dependency:
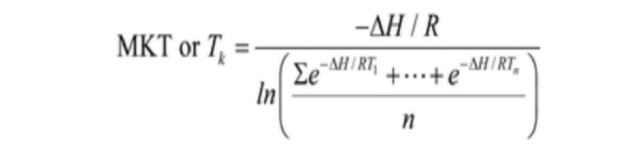
Where:
- ΔH = The heat of activation for the degradation reaction; a good approximation is 83.144 kJ/mol
- R = 8.3144 × 10-3 kJ/mol ·K (universal gas constant)
- T1 = Value for the temperature, in degrees Kelvin, recorded during the first time period, which expresses in minutes, hours, days, or weeks
- Tn = Value for the temperature, in degrees Kelvin, recorded during the nth time period
- n = Total number of storage temperatures recorded during the observation period
When applying MKT to evaluate Controlled Room Temperature (CRT) Stability Chamber excursions, it is recommended to use not less than 30-days of temperature data for the observation period, which is consistent with the USP 30-day recommendation for calculating MKT.12,13,14,15
For example, using guidance similar to what is found in U.S. Pharmacopeia (USP), General Chapter <659> for CRT, MKT may be used during a stability chamber excursion period, provided:
- MKT does not exceed 27°C;
- Excursion range is between 15 and 30°C;
- Transient spikes NMT 40°C.
As an example of a low-temperature excursion, consider the case where the temperature falls to 18°C for 23 hours, there were no transient spikes, and the MKT is calculated to be 24.8°C, there should be no impact to the product stability samples stored in the chamber during the excursion. The MKT calculation and rationale should be included in the deviation investigation and stability data package.
In a similar case, where the temperature in the CRT chamber increases to 29°C, and the calculated MKT exceeds 27°C, the MKT cannot be used to determine if there is any impact risk to the stability samples. Data from accelerating aging studies and information from previously conducted stress testing will be used to further confirm the risk along with much other information that may be available to structure a risk assessment and establish follow-up actions such as the following.
Leverage Data from Accelerated and Stress Studies
Forced degradation studies can identify stability risks. Per ICH Q1A(R2), stress testing should include the effect of temperatures (in 10°C increments above that for accelerated testing), and humidity (e.g., 75% RH or greater). Examining degradation products under temperature and humidity extremes helps establish the degradation pathways and suitable stability-indicating analytical procedures.
For the case, where a 40°C accelerated aging chamber had drifted up to 48°C and remained there for 36 hours, if a previous stress study had been conducted at 50°C for up to a week for each type of product sample contained in the chamber, we would have a good idea as to the type of degradation that the samples would experience, in addition to knowing how the critical quality attributes might be impacted. Having such data available prior to experiencing a chamber excursion will prove to be invaluable.
While this article is emphasizing temperature and humidity excursions, one should also be mindful of the photostability of the drug substance and drug products. Many stability samples may be stored in the primary packages in the stability chambers, and they may be exposed to light. If degradation occurs, any impurities should be identified as originating from storage (aging) or light excursions. Therefore, forced degradation studies can play a key role in the investigation of a chamber excursion, and provide additional data to evaluate the risk impact to sample integrity.
Adding Additional Time to Stability Sample Pull Schedule
When the temperature decreases for longer than 24 hours, the most conservative approach requiring minimal justification is to add additional time to the scheduled pull dates for each of the impacted stability studies.
Consider the case of a (50°C) chamber used for accelerated aging of medical devices. Figure 2 shows a temperature and humidity excursion at 10 PM on the evening of December 23rd and which was resolved by 2 PM on December 24th (approximately 16 hours). A subsequent excursion occurred early on Christmas morning at 12:40 AM and lasted until 9:30 AM on December 31st (approximately 153 hours). The lowest temperature recorded during this second excursion was 31.4°C, and the highest was 51.8°C, while the lowest humidity was 14% RH, and the highest was 74% RH.
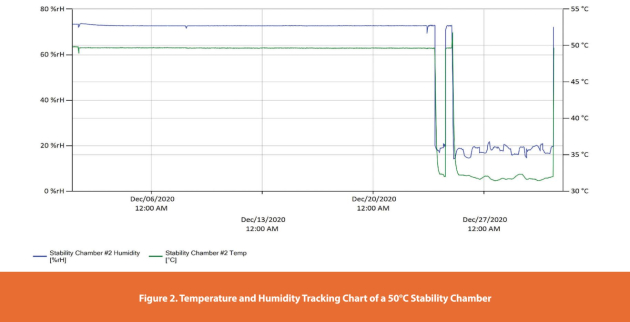
In this case study, since the first excursion lasted less than 24-hours, the event was noted with no additional action was required. The second excursion lasted 153-hours (~7 days), so an investigation was necessary. Since the temperature and humidity dropped outside of the range for such an extended time, there would be reduced degradation kinetics based on the Q10 factor for the samples. Based on the guidance, this excursion could be detailed in the stability reports for all affected product samples; an additional 7 days (153 hours) should be added to each subsequent sample pull to make up for the 153-hour temperature excursion. However, the excursions should be considered as part of the overall situation. If this is the first failure of this particular chamber, then it could be equipment-related; however, if it is a repeat occurrence, then the previously established root cause should be re-evaluated to determine if the additional effect is the cause.
The impact of the humidity excursion may also need to be assessed if the samples are in semi-permeable or permeable packaging. For stability studies evaluating the effect of humidity on a sensitive product, it is important to psychrometrically calculate equivalent water vapor (ppm) at various temperatures and relative humidities. For the lowest reading of 31.4°C and 14% RH, the water vapor concentration is 6397 ppm at sea level, while at 51.8°C, and 74% RH, the water vapor concentration is 109,370 ppm. Converting to water vapor concentration (in ppm) allows the impact of any hydrolytic degradation kinetics to be evaluated.
Testing Extra Samples for Additional Data
Extra stability samples stored at matching conditions are helpful to conduct investigations of stability-related issues, and even to perform testing past the final time interval if desired. Depending on the nature of the stability study, sample size, cost, and availability of samples, it is recommended that a strategic number of additional samples (up to 100%) is placed in each chamber at the start of each study. All samples, including these extra investigational samples, must be accurately accounted for in the chamber inventory.15
In the event of a temperature/humidity excursion, extra samples can be pulled and tested to determine the impact of the excursions without disturbing the protocol’s defined pull samples. The data from the investigatory samples will be part of an investigation plan for the chamber and will be included in the detailed report describing the risk of impact on the samples. The testing can include critical-to-stability test methods from the pre-approved protocol and/or fewer routine tests which are useful to indicate temperature/humidity-induced oxidation/hydrolysis risk.
Documenting Chamber Excursions
Each stability chamber will require a log where qualification, calibration, and maintenance information are documented. All excursions must also be documented as a deviation in the corresponding chamber’s log, regardless of the length of the excursion. However, the SOP should define the difference between routine activities such as opening the door, electrical surges, and an excursion to properly document these incidents (Table 2). An investigation should be initiated when the excursion exceeds 24 hours or if the magnitude of deviation is critical. For example, one may note a series of short-term, high-magnitude excursions that may become a critical impact on the materials with phase changes near the excursion. It is also helpful to track all excursions and periodically review this list to identify any potential chamber performance issues requiring updates to the preventative maintenance program before additional excursions occur.

Establish a Procedure to Handle Chamber Excursions
A stability chamber Standard Operating Procedure (SOP) must include a section on how to handle and document chamber excursions. It should provide specific steps leading to what data are to be evaluated and which actions are to be taken for the investigation. When an excursion exists, the first activity should be to mitigate further impact to any stored samples. This is followed by performing the investigation to disposition the samples, and finally determining the root cause.
All deviations should be documented. When frequent deviations occur, the investigation should address these deviations as part of the equipment performance maintenance. Chamber calibration, qualification (IQ, OQ, and PQ) and maintenance documentation are also critical to the investigation. Risk evaluations should also be done when multiple excursions occur. The magnitude of an excursion can instantaneously degrade samples when a phase change is crossed.
Stress Test/Forced Degradation studies should be conducted before formal stability studies are initiated to understand the stability profile of the products. As noted previously, this data can objectively bound the risk from an excursion.
It is critical to trend the chamber performance frequently. This activity must be included as part of chamber routine maintenance. Companies should also develop a contingency disaster plan for stability chambers to handle situations when the excursion exceeds 24 hours and/or the temperature and humidity changes excessively. In the case of a chamber malfunction that cannot be repaired within 24 hours, it is advisable that backup qualified chambers are available so that samples can be transferred temporarily. These backup chambers can be located on-site or at an alternate site that has sufficient capacity to accommodate the stability samples.
Conclusion
Properly maintained stability chambers are a critical component of any well-run Stability Program. Regardless of the diligence, a temperature and or humidity excursion will occur at some point. Several different approaches can be utilized to assess the impact of the excursion on the stability samples. Excursions lead to changes in the chemical or physical (and even geometric) characteristics of the samples, depending on the duration and magnitude. There are tools such as the Mean Kinetic Temperature (MKT) concept, stress testing/forced degradation studies, accelerated condition studies, and additional post-excursion extra sample testing that can be used to evaluate the impact risk of the excursions on the quality and integrity of the samples stored in that chamber. Once the impact risk is understood, mitigation steps such as increased aging or protocol testing enhancements can be determined.
Acknowledgments
The authors express their gratitude to the following colleagues for their time and effort to provide outstanding technical peer reviews: Josh Hansen, Hollister Incorporated; Desmond Hunt, Ph.D., United States Pharmacopeia; Laure Larkin, M.S., Ethicon; Tony Mazzeo, Ph.D., Bristol-Myers Squibb; and John O’Neill, M.S., Stabilityhub.com.
References
- Food and Drug Administration. Guidance for industry Q1A (R2) Stability Testing of New Drug Substances and Products, Food and Drug Administration, Rockville, MD. Available at https://www.fda.gov/media/71707/download.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Tripartite Guideline: “Stability Testing of New Drug Substances and Products” Q1A(R2), Available at: https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf
- World Health Organization, “Stability Testing of Active Pharmaceutical Ingredients and Finished Pharmaceutical Products”, Working document QAS/17.694, Available at: https://www.who.int/medicines/areas/quality_safety/quality_assurance/stblty-testing-APIsandFPPS-QAS17-694_12012017.pdf
- Advanced Botanical Consulting & Testing Inc dba ABC Testing, FDA Warning Letter, June 4, 2019. Available at https://www.fda.gov/inspections-compliance-enforcement-and- criminal-investigations/warning-letters/advanced-botanical-consulting-testing-inc-dba-abc-testing-572991-06042019.
- Ercros S.A. FDA Warning Letter, June 20, 2012. Available at https://www.fdalabelcompliance.com/letters/ucm310963
- Jacobus Pharmaceuticals, FDA 483, 2009. Available at https://www.fda.gov/media/88766/download
- Aarti Drugs Limited Warning Letter, July 30, 2013. Available at https://app.gxp-services.net/external/warning_letters/13%2007%2030%20Aarti.pdf
- Anderson, C, Seevers, R. “The Use of Mean Kinetic Temperature to Aid Evaluation of Temperature Excursions for Controlled Cold Temperature Drugs: Proper and Improper Application.” AAPS Webinar, July 9, 2020.
- Seevers, R, Hofer, J, Harber, P, Ulrich, D, and Bishara, R. “The Use of Mean Kinetic Temperature (MKT) in the Handling, Storage, and Distribution of Temperature Sensitive Pharmaceuticals,” Pharmaceutical Outsourcing, May/June 2009.
- Lucas, T, Bishara, R, and Seevers, R. “A Stability Program for the Distribution of Drug Products,” Pharmaceutical Technology, July 2004.
- Haynes, J. “Worldwide Virtual Temperatures for Product Stability Testing”, J. Pharm. Sci., 60:927-929, 1971.
- United States Pharmacopeia, General Chapter <659>, “Packaging and Storage Requirements”, USP43-NF 38 2S.
- United States Pharmacopeia, General Chapter <1079.2>, “Mean Kinetic Temperature in the Evaluation of Temperature Excursions During Storage and Transportation of Drug Products”, USP43-NF38 2S.
- Center for Healthcare Supply Chain Research. Factbook: The Facts, Figures, and Trends in Healthcare (2015–2016). 86th ed. Arlington, VA: Center for Healthcare Supply Chain Research; 2016:15.
- Huynh-Ba K., Ed., Handbook of Stability Testing for Pharmaceutical Products: Regulations, Methodologies, and Best Practices. New York, NY: Springer Science & Business Media; 2009.
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Tripartite Guideline: “Quality Risk Management, Q9. Available at: https://database.ich.org/sites/default/files/Q9%20Guideline.pdf
Author Biographies
Chris Latoz is Stability Manager for Hollister Incorporated, a company that develops, manufactures, and markets healthcare products worldwide. Chris has extensive experience in the medical device industry in R&D, materials testing and characterization, and stability. Chris is a member of American Association of Pharmaceutical Scientists (AAPS), where he currently serves as Vice-Chair for the Stability Focus Group. Chris can be contacted at [email protected].
Kim Huynh-Ba is the Managing Director of Pharmalytik, LLC, a consulting and training organization. Kim is a member of the USP Council of Experts (2015-2025), where she chairs the Small Molecules 4 Expert Committee. Kim is an Adjunct Professor at Temple University-School of Pharmacy and Illinois Institute of Technology (IIT), teaching Quality Audit, ICH quality guidelines, Good Manufacturing Practices. She is the editor of multiple book volumes, including the “Handbook of Stability Testing in Pharmaceutical Development: Regulations, Methodologies, and Best Practices” (2008), “Pharmaceutical Stability Testing to Support Global Markets” (2010), and the upcoming “Analytical Chemistry: An Introduction to the Pharmaceutical GMP Laboratory,” (in press 2021). She can be contacted at [email protected].
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!