Introduction
Conventional pharmaceutical manufacturing is based on batch manufacturing processes which are robust and have been producing high quality materials for decades. However, batch manufacturing processes contain numerous, disconnected steps; exhibit significant scale changes throughout development and commercialization; and typically involve product testing after manufacturing is finished.

With Continuous Manufacturing (CM), processes could be designed such that there is constant material flow in and out of the process. While CM is already established in several industries including petrochemical, food and polymer industries, it is relatively new in the pharmaceutical industry. CM use in the pharma industry is growing as it provides several opportunities for improvements. A few pharmaceutical unit operations are already continuous, by design. Examples include tablet compression operations, milling operations and others. However, adoption of CM in pharmaceutical manufacturing impacts to a large extent the integration of individual unit operations, and such integration allows for a number of business and technical opportunities with CM that are not allowed for with conventional batch processes.
This integration of unit operations with CM is very different from established batch manufacturing operations where a significant amount of time may elapse in between subsequent unit operations. For example, dispensing operations may take place days or weeks ahead of the start of manufacturing, and days or weeks may also elapse between subsequent processing steps until the final product is produced with batch processes. On the other hand, CM allows for integration of several unit operations such that processing across the entire manufacturing train could be taking place at the same time. In that manner, input raw materials can be continuously fed into the CM train, all unit operations are integrated and operational, and finished product is continuously produced out of the system.

This integration also allows for new methodologies to be applied to some of the unit operations to allow for such integration. The most prominent examples of this are the dispensing and blending operations which leverage newer equipment designs and allow for improved process controls than their counterparts in conventional batch manufacturing operations.
It is a misconception that CM is only appropriate for large volume products. CM does not mean manufacturing operations need to operate continuously over very long periods of time. There are business and technical advantages to leveraging CM operations even for small volume products. Factors including the integration of the unit operations, the opportunities to leverage new technologies for some unit operations, the reduction or elimination of some of the notorious manufacturability risks that are historically observed with conventional batch processes, the ability to reduce the equipment footprint, and the opportunities to apply advanced modes of process monitoring and controls collectively enable CM to be a very disruptive and beneficial platform for pharmaceutical operations, regardless of the expected product volume. Some of these benefits include ability to conduct faster and more efficient development, the ability to reduce technical risk by conducting development studies at the same equipment size as commercial production, the ability to enable higher supply chain agility, and the ability to design appropriate controls into the CM system rather than the industry historical standard of relying mostly on testing quality at the end of production.
One important aspect of those benefits that are allowed through CM is the fact that CM can enable faster approaches to develop and commercialize products. This is the focus of the remainder of this article and will be highlighted using drug product CM examples.
Background on Drug Product CM Operations
The three main manufacturing platforms for solid oral drug products are direct compression, roller compaction, and wet granulation. All three platforms are possible with CM. However, CM allows for process simplification due to the elimination or reduction of segregation or flow related manufacturability risks which are historically observed with batch manufacturing processes. This leads to higher utilization of simpler manufacturing platforms with CM like Continuous Direct Compression (CDC). Therefore, CDC is currently the most commonly used CM platform in the pharmaceutical industry, as evidenced by the number of currently approved products with CDC.
Drug product development is expected to be more efficient with CM. As will be discussed, with a higher utilization of simpler manufacturing processes (e.g., CDC), the use of simpler formulations, and reduction or elimination of scale up risks, the time from development to commercialization can be reduced.
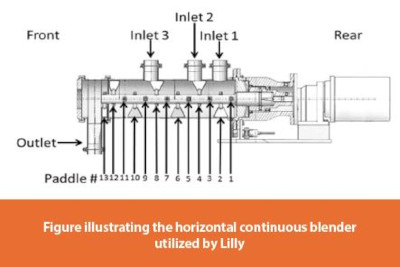
Figure 1 illustrates Lilly’s CDC unit. Lilly designed, optimized, and implemented a CDC process for drug product manufacturing which consists of three major unit operations: Loss-In-Weight (LIW) feeding of raw materials via powder screw feeders, continuous blending of these material streams using a horizontal paddle mixer, and tablet core compression using a rotary tablet press. The process is simple by design and is assembled in a vertical stack, utilizing gravity to promote consistent powder flow. An automated distributed control system is used to carry out the batch recipe, communicate with and collect data from all sources, and execute the control strategy. Multiple layers of control such as measurements from equipment soft sensors and spectroscopic Process Analytical Technologies (PAT) are used throughout production to monitor the health of the process and detect any potential upsets. These elements along with predefined criteria for product collection, diversion, and/or isolation, enable a state of control to be maintained and provide a real-time assurance of quality.1 A unique aspect of the continuous process is the ability to integrate all these features into one wholistic system, including not only the sophisticated equipment and controls, but additionally the sensing and characterization tools, the information technology and data analytics, and also human factor considerations. This modernized approach allows for quicker execution and analysis of experiments in development and facilitates a deep understanding of the process.
Using CM to Improve Efficiency and Speed the Development Process
Continuous manufacturing enables use of both simpler and a wider range of drug product formulations while using simpler manufacturing processes. It is not uncommon for pharmaceutical formulations to have poor physical properties that present risks such as inconsistent fl ow and segregation. In batch processing, these risks are exacerbated where powders are stored and transferred in bins. Often during transfer steps, forces are applied to the particles creating interparticle movement.2 This can lead to the concentration of particles with specific properties such as size, density, or shape and thus affect performance of the downstream operation and potentially impact product quality. In addition, often large quantities of material create a load or head pressure within the bin, causing stagnation and resistance to flow. Such formulations often require additional processing steps such as granulation to enhance the physical properties for a robust operation. This however adds processing complexity, cost, and time. In effect, time, effort, and money is spent to design around complexities that are brought about by the inherent nature of batch processing. In a CM process, on the other hand, powders are continuously in motion from the point of feeding to compression, thus preventing stagnation and de-aeration points in the process.

Example 1 (segregation): To illustrate this further, a formulation which is prone to segregation and requires a dry granulation (roller compaction) step when utilizing a batch process was processed using CDC (without any granulation step). The results shown in the figures below demonstrate that the CDC process can produce equivalent product quality compared to the batch roller compaction process even with the troublesome formulation. This is illustrated by analyzing stratified samples for potency and content uniformity throughout both the batch and continuous processes. In addition to mitigating the risk of segregation, by eliminating the roller compaction step, the compactibility of the formulation was significantly improved, resulting in increased tablet breaking forces when using the CDC process with the same formulation. This is particularly important for developing formulations for new chemical entities where a portion of the compactibilty of the blend is not “lost” intra-granularly through use of roller compaction. Thus, this allows the opportunity when developing new formulations for use of simpler formulations and in less time and with less effort since there is no need to invest significant resources and materials to investigate and optimize the interplay of formulation compositions and densification operations in order to achieve acceptable tablet tensile strengths while reducing segregation risks. CM enables use of simpler operations with simpler formulations and allows for acceptable uniformity while providing wider compactibility margins.
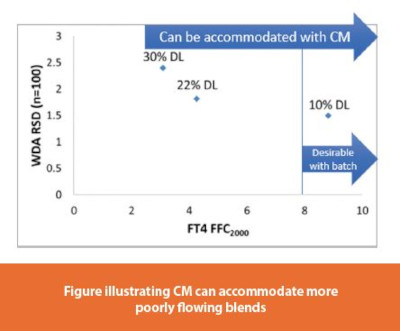
Example 2 (flow): Furthermore, CM accommodates more poorly flowing formulation blends compared to batch manufacturing. When using a batch direct compression process, it is desired for the powder blend to have a relatively high flow index value in order to control tablet weight variability.3 However, when using the CDC process, formulations with a wider range of acceptable flow properties have been demonstrated to process robustly. As a result, the API concentration or drug load in direct compression formulations may be able to be increased with CM well beyond the ranges that historically could have been achieved with conventional batch processing. Maximizing drug load is ideal and has direct benefit for the patient as smaller dosage forms or fewer dosage units per dose can be achieved. The figure above, right illustrates an example where the drug load for a given formulation was successfully increased from 10% to 30% when transitioning from batch processing to CDC. In this case a much lower formulation blend FFC value was accommodated while still maintaining tablet weight control in the desired range (e.g. < 3% RSD).
With these powder flow risks reduced, a wider range of formulations can be used via CDC leading to an increased use of the simpler continuous processes. These efficiencies benefit both development, as discussed, and manufacturing through increased asset utilization of a leaner process with a smaller footprint and a shorter cycle time.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
CM Enables Use of Platform Understanding Across Multiple Molecules to Speed Development and Commercialization
CM also enables the use of platform understanding across multiple molecules to further drive the efficiency of the development process. For example, a library of LIW feeding performance data can be generated for commonly used CDC formulation components across equipment confi gurations and control settings. The use of a LIW feeding system level ratio controller has also been demonstrated to reduce material concentration variability in the dispensed powder stream into the blender and this control architecture can be replicated across projects.4 In addition, the horizontal mixer design including mechanical setup and operational conditions has shown to be robust across multiple challenging formulations and a low risk to impact product critical quality attributes (CQAs).5 Various predictive process models and PAT techniques have also been utilized to generate platform-based understanding on topics such as characterizing residence time distributions (RTD), material traceability and diversion strategies, and the handing and propagation of disturbances. These experiences have led to platform wide development strategies that can be leveraged across projects and subsequently reduce the number of required experiments and thus shorten development timelines. A science and risk-based development approach is then leveraged to help identify and focus development studies on the remaining residual risks that are relevant to the new molecules. In this manner, through use of a common, preferred CM platform across multiple molecules, use of process modeling and understanding, and thorough raw material characterization, a sponsor may leverage a science and risk-based approach with focused experimentation and multi-molecule platform understanding for justifying control strategies for a given asset instead of recreating all development data for every new asset.
Lower Scale Up and Technical Transfer Risks by Developing at Scale
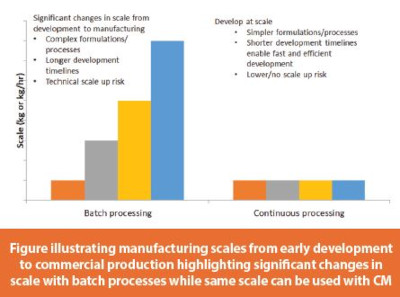
Batch manufacturing processes are almost synonymous to “scale-up”. It is typical to initiate early development studies at very small scales (mainly due to time and material availability limitations), and then the scale of the batch manufacturing process may increase several times throughout the development and commercialization process. It is common to “lock” formulation compositions and select the manufacturing process platform during clinical stages of development, at scales that are much smaller than the eventual intended commercial scales because it is important to maintain bridging between products and processes used in pivotal registration studies and primary stability studies to those intended for commercial production. Due to the significant changes in scales from development to commercial scale, this in turn forces the formulation scientists and process engineers to conduct many experiments to understand scale-up risks (e.g., overlubrication) and in many cases, leads to the use of more complex formulations and more complicated manufacturing process platforms in order to “build in robustness” into the product and process to mitigate any potential issues upon final scale-up to commercial scales. This typically leads to longer development timelines and more expensive development as scale-up introduces a level of risk and variability and therefore must be well understood and studied in development. Furthermore, demonstration of the product and process robustness may need to be proven at several points during the overall development process, adding more time and cost. Even with all these efforts, it is inevitable that scale-up risks may still be encountered for some products upon final scale-up to commercial scale. Such issues can be costly as they may occur very late in the commercialization plans close to process validation or product launches.
With CM, a completely different paradigm is possible. While early formulation development studies could commence at very small lab scales, some, or a majority of development processes to confirm or finalize formulation composition and/or processing details can be executed at commercial equipment size. This “develop at scale” paradigm is tremendous as the development scientist can observe early in development how the intended formulation and process would perform at commercial equipment size. This in turn can enable use of simpler formulations and processes as the ambiguity of future scale-ups would be eliminated. This results in shorter development timelines, and leads to lower technical transfer risks, especially in cases where the CM platform is very well understood from previous assets or in cases where the development CM rig is the same or very similar in size and design to the commercial CM rig. Thus, little or no scale-up effort is required, creating a simpler, streamlined technology transfer when transitioning a product from development to GMP sites through the use of consistent equipment, automation, and controls. The commercial CM rig could then be used to produce larger batch sizes, if needed, by running the process longer or increasing the throughput rate.6
It is not uncommon for significant tablet compact strength variability to be observed as a function of scale when using a batch process. The figure below demonstrates this by comparing tablet breaking force across equipment scales. This risk will be reduced or eliminated with CDC since development and manufacturing utilize replicated equipment trains, thus reducing the potential for equipment scale to influence product attribute variability.
Conclusions
With improved efficiency of CM processes, shortened development timelines can be achieved, thus speeding time to commercialization. As discussed in this article, this can be achieved through the use of a simpler, integrated process which can leverage platform understanding across multiple molecules and apply simpler formulations. In addition, development can be performed at identical equipment scales, thus eliminating or reducing scale up risks and enabling simple technology transfer to GMP sites through replicated equipment, automation, and controls.
References
- Almaya A, De Belder L, Meyer R, et al. Control Strategies for Drug Product Continuous Direct Compression – State of Control, Product Collection Strategies, and Startup/Shutdown Operations for the Production of Clinical Trial Materials and Commercial Products. Journal of Pharmaceutical Sciences. 2017;106:930-943.
- Liu G. Understanding and minimizing powder segregation. Powder Bulk and Engineering. May 2018.
- Divya S, Ganesh GNK. Characterization of Powder Flowability Using FT4 – Powder Rheometer. J Pharm. Sci. & Res. 2019;11(1):25-29
- Hanson J. Control of a system of loss-in-weight feeders for drug product continuous manufacturing. Powder Technology. 2018;331:236-243.
- Roth WJ, Almaya A, Kramer TT, Hofer JD. A Demonstration of Mixing Robustness in a Direct Compression Continuous Manufacturing Process. J Pharm Sci. 2017;106(5):1339-1346.
- Lee SL, O’Connor TF, Yang X, et al. Modernizing Pharmaceutical Manufacturing: from Batch to Continuous Production. J Pharm Innov. 2015;10:191-199.
Author Biographies
Ahmad Almaya is Director of Global Regulatory Affairs at Lilly, where he is responsible for CMC regulatory activities supporting global regulatory submissions for Lilly synthetic molecule development projects and commercial products from US manufacturing sites. Ahmad received his B.S. in Pharmacy from The University of Jordan, Ph.D. in Pharmaceutics from The University of Iowa, and an Executive Certificate in the Business of Life Sciences from Kelley School of Business, Indiana University.
Joshua Hanson is an Associate Senior Consultant Engineer at Eli Lilly working in the Synthetic Molecule Design and Development organization within Lilly Research Labs. Josh’s focus as a drug product development engineer is to apply modernized technologies and commercialization approaches to new medicines including continuous manufacturing and help bring those medicines to patients. Josh received his B.S. in chemical engineering from Purdue University in 2007 and a Professional Engineering License in 2014.