Department of BioMolecular Sciences
Introduction
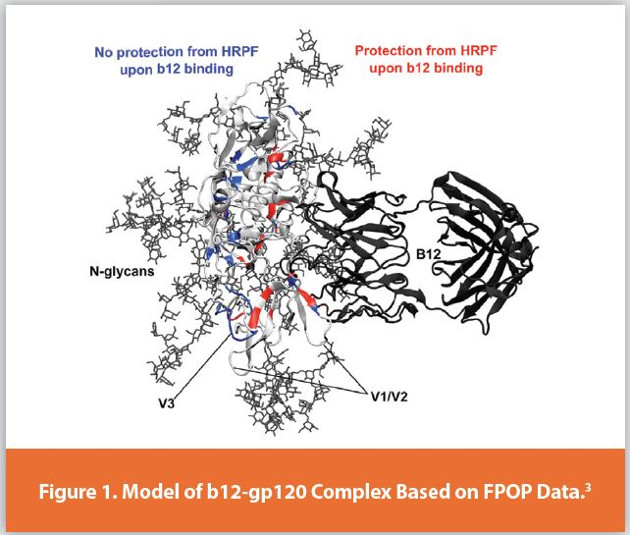
Protein higher order structure (HOS) analysis is one of the most complicated steps in biopharmaceutical analysis. Hydroxyl radical protein footprinting (HRPF) is a technology developed over the past three decades for characterizing protein higher order structure using mass spectrometry.1,2 Proteins are exposed to freely-diff using hydroxyl radicals in solutions, and these hydroxyl radicals modify amino acid side chains based, in part, on their solvent accessibility. The amount of side chain oxidation is measured by proteolysis followed by liquid chromatography coupled to mass spectrometry (LC-MS). By comparing the footprint of a protein sequence in two different structural states (e.g. two different formulations; ligandbound vs. ligand-free; glycosylated vs. deglycosylated; etc.), changes in the protein topography can be detected and localized based on changes in the amount of oxidation of a given section of a protein. This information can be used to determine antibody epitopes or ligand binding sites, regions of instability or aggregation interfaces, proteinprotein interaction surfaces, sites of allostery, or answer numerous other HOS questions with no theoretical limitations on analyte size and tremendous flexibility on sample matrix. As one example, HRPF by FPOP was used to identify the broadly neutralizing b12 antibody epitope in fully glycosylated gp120 of HIV (Figure 1).3 This represented Figure 1. a very challenging problem in HOS characterization, due to the flexibility of the gp120 variable regions and the high N-linked glycan content (~50% of the total glycoprotein mass). While many benchtop methods for HRPF have been reported, they all share similar concerns that must be addressed in experimental design: maintaining native structure throughout the labeling process; preventing secondary oxidation; measuring and compensating for differential radical production and scavenging in your sample; and understanding the limitations of data analysis and interpretation. This article discusses these critical concepts along with their impact on HRPF results, focusing on a popular benchtop method for HRPF radical generation: Fast Photochemical Oxidation of Proteins (FPOP).4 When these factors are understood and controlled, HRPF generates unique and sensitive HOS information in a highly flexible platform, and adds considerable value when integrated into existing HOS analysis workflows.
Controlling Oxidation in HRPF Samples
It has been established that the apparent rate of oxidation of an amino acid side chain by hydroxyl radicals in HRPF depends upon the chemical nature of the amino acid side chain (it’s chemical structure5,6 and its amino acid sequence context)7,8 and the accessibility of the side chain to hydroxyl radical diffusing in dilute solution.8-10 However, the stable oxidation products generated by reactive oxygen species, including the hydroxyl radicals used in HRPF, alter the biophysical properties of the side chains they affect, often making hydrophobic residues considerably more hydrophilic. These modifications can alter protein conformation. If researchers continue to label oxidatively unfolded proteins, they will introduce artifacts into the topographical analysis and confound data interpretation. Therefore, it is necessary to carefully control the conditions and understand sources of protein oxidation in HRPF experiments.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
HRPF-Induced Conformational Changes
There are two general means to prevent the introduction of these labeling artifacts into HRPF results: limiting the extent of labeling to prevent the introduction of oxidatively unfolded proteins through some means of measuring HOS changes during labeling (e.g. circular dichroism spectroscopy,11 oxidation kinetics analysis),8,12 or complete the oxidation chemistry on a shorter timescale than large scale protein motions to prevent any labeling of an oxidatively unfolded conformation.4,13,14 It is the ability to do the latter in a benchtop format that has made FPOP attractive for HRPF analysis. Figure 2 shows a typical modern FPOP bench setup. Samples are mixed with hydrogen peroxide and pushed through a capillary through the path of a pulsed KrF excimer laser, emitting light at 248 nm. The KrF laser is masked with a 1 mm aperture which limits the length of capillary illuminated by the laser, correcting for beam jitter and beam expansion/contraction at different pulse energy. When the laser pulses, it illuminates a volume of sample within the capillary for tens of nanoseconds with UV light. The UV light photolyzes the hydrogen peroxide, generating a shortlived high concentration of hydroxyl radicals for HRPF. The lifetime of the hydroxyl radicals is further controlled by the addition of a radical scavenger, which ensures the hydroxyl radicals are consumed on the order of ~ 1 μs4 (although protein radicals generated and other radical species persist longer).14,15 The illuminated volume of sample is then pushed out of the path of the excimer laser and the next volume is illuminated, with an unilluminated volume in between to correct for laminar flow effects and axial diffusion of sample.4 Empirical results have shown that, when the proper exclusion volume (usually 15% for most flow regimes) and the proper hydroxyl radical scavenging conditions (usually ~20 mM glutamine or an equivalent scavenging capacity) are used, even oxidatively sensitive proteins are labeled faster than they can unfold.4,8
Secondary Oxidants
While HRPF uses the apparent rate of oxidation by hydroxyl radicals to measure changes in protein topography, hydroxyl radicals are not the only protein oxidants used or generated in HRPF. Oxidation of the protein by these secondary oxidants result in many of the same protein oxidation end products as hydroxyl radicals, and can result in artifactual results that obscure protein HOS information. In FPOP, proteins are first mixed with hydrogen peroxide. Hydrogen peroxide alone is capable of two-electron oxidation of proteins, most commonly at sulfur-containing amino acid side chains such as methionine and cysteine. Even in water radiolysis-based experiments that do not use hydrogen peroxide, hydrogen peroxide is a common product of the recombination of two hydroxyl radicals. Hydroxyl radicals will also generate other secondary oxidants such as superoxide and singlet oxygen from dissolved molecular oxygen and other constituents of the sample.16 Additionally, hydroxyl radical-mediated oxidation of certain amino acid side chains (most notably aliphatic side chains) can result in mixed peptidyl hydroperoxides that can participate in two electron oxidation reactions similar to hydrogen peroxide.17 While these secondary oxidants are not nearly as reactive as the hydroxyl radical, all of them can oxidize the protein analyte given sufficient time.

In FPOP, protein residence time in hydrogen peroxide is limited to minimize peroxide-induced oxidation of cysteine and methionine. Proteins are either mixed with hydrogen peroxide immediately prior to FPOP illumination or, in some cases, hydrogen peroxide is mixed with the sample on-line.18,19 Immediately after illumination, sample is dispensed into a quenching vessel containing catalase, a highly efficient enzyme that converts hydrogen peroxide into water and molecular oxygen. Together, these steps limit the exposure of the protein to hydrogen peroxide; however, other secondary oxidants present at lower concentrations (peptidyl hydroperoxides, superoxide, singlet oxygen) are not catalytically degraded by catalase. To eliminate these secondary oxidants, a scavenger containing a thioether (usually methionine amide) is also added to the quenching vessel with catalase at millimolar concentrations. The thioether is highly reactive to both one- and two-electron oxidation, and quenches the remaining secondary oxidants. With proper control, secondary oxidation can be limited to undetectable levels,16 but the use of unilluminated controls (wherein the sample is exposed to FPOP conditions without illumination by the excimer laser) are crucial to ensure that proper secondary oxidation control is achieved.

Radical Dosimetry and Scavenging Compensation
One of the major hurdles to the inclusion of FPOP and other HRPF technologies to a biopharmaceutical environment is the question of reproducible radical exposure. The amount of hydroxyl radical that is available to react with the analyte protein in an FPOP experiment is subject to a wide array of factors, only some of which are under the direct control of the experimenter: photon density of the UV laser pulse; optical properties of the fused silica capillary; hydrogen peroxide concentration; and, most notably, the radical scavenging properties of other molecules in the sample. This question of radical scavenging is particularly important in biopharmaceutical analysis. The same high oxidation potential of the hydroxyl radical that makes it such a broadly useful label for protein topographical analysis also means it will react with almost any organic molecule in solution with the protein. In biopharmaceutical formulations, these molecules often include buffers, ligands, excipients, carrier proteins, etc. In the presence of a higher concentration of radical scavenging capacity, a protein with the exact same topography will show significantly reduced oxidation simply due to the fact that the analyte encounters fewer hydroxyl radicals. Since the differential HOS analysis to be carried out is often examining the structural eff ects caused by the addition of one of these organic molecules (e.g. ligand-bound versus ligand-free), understanding and controlling the radical scavenging properties of the sample is critical to maintaining robustness, reproducibility, and proper analysis of the HRPF data.

Adenine Radical Dosimetry
The common method for measuring the effective amount of hydroxyl radical delivered to the analyte is the use of a hydroxyl radical dosimeter: a molecule used as an internal standard that alters its properties upon reaction with hydroxyl radicals. Several different methods of hydroxyl radical dosimetry have been published for FPOP,2,20,21 but perhaps the simplest to implement is the use of adenine as an internal dosimeter. Adenine is moderately reactive with hydroxyl radicals, and can be used in place of commonly used hydroxyl radical scavengers such as glutamine. Adenine has a strong UV absorbance band at 265 nm; however, adenine that has reacted with hydroxyl radicals has significantly lower absorbance at 265 nm (Figure 3). As the adenine competes for the same pool of hydroxyl radicals as the protein analyte, changes in the absorbance of the adenine dosimeter directly reflect changes in the effective concentration of hydroxyl radicals (i.e. the amount of radical generated minus the amount reacting with other scavengers in solution).22,23 With the use of an inline UV spectrophotometer as shown in the optical bench in Figure 2, the effective hydroxyl radical dose can be monitored in real time.24 Not only does this allow for real-time trouble-shooting, but the experimenter can compensate for different scavenging capacities of the sample in real-time.
Dosimetry-Based Compensation
A kinetic simulation of the compensation process is shown in Figure 4. A dosimeter response is measured in the reference sample, and set as a target (e.g. 49% oxidation of a 1 mM dosimeter, black trace). Upon addition of a scavenger excipient (1 mM histidine in the simulation), the dosimeter response decreases (blue trace) along with the amount of protein oxidation in a proportional manner (cyan). When the amount of hydroxyl radical generated is increased to give an equivalent 49% yield of oxidized dosimeter in the sample with 1 mM histidine as achieved with 1 mM hydroxyl radical in the absence of histidine (in this case, 1.82 mM •OH is required, red trace), the amount of protein oxidation that occurs similarly becomes identical (magenta).
We have termed the process of tailoring radical yield based on dosimeter response “compensation”. In FPOP, compensation can be achieved by altering hydrogen peroxide concentration and/or UV light fluence impinging upon the sample to alter hydroxyl radical production, achieving equivalent dosimeter response. While this strategy can be used to “tune down” conditions to the level attained in this highest radical scavenging background, this strategy results in a loss of information: resulting analyte proteins have fewer oxidation events per molecule, resulting in less topographical information. More usefully, experimenters can “tune up” conditions in the sample with higher radical scavenging capacity to the desired level without compromising the information generated. Using this real-time compensation, the radical scavenging capacity of different formulations (buffers, excipients, etc.) can be overcome. This ensures that differences in the HRPF footprint will reflect differences in protein HOS (Figure 5).24

Bringing FPOP to Industrial Settings
While FPOP has largely remained confined to academic and government laboratories, the technique has recently been used to a limited degree in industrial biopharmaceutical settings as recently reviewed by Garcia and coauthors.25 Several hurdles that originally limited the flexibility and reproducibility of FPOP have been addressed: radical dosimetry, higher laser fluence through high energy lasers8 or cylindrical lenses2 to overcome radical scavengers commonly found in biotherapeutic formulations, and more detailed understandings of factors underlying FPOP precision26 and prevention of secondary oxidation.16,17,27 The stability of the protein oxidation products makes FPOP compatible with the wide variety of sample workup processes that are commonly needed for efficient analysis by bottom-up mass spectrometry (e.g. deglycosylation, disulfide reduction, thermal denaturation, etc.) without concerns for back-exchange and without theoretical limitations on analyte protein size or dynamics. The information on protein surfaces generated by FPOP places it in a unique niche in options for HOS analysis, while its sensitivity to changes in HOS is greater than some techniques popularly used in biotherapeutic HOS analysis.28 However, the technique remains technically challenging, both experimentally (handling and maintenance of unembedded Class 4 UV lasers) and in terms of data analysis. Data analysis software has progressed considerably,29,30 but remains in its early stages and still usually requires manual auditing for accurate quantification of oxidation. As advancements progress to simplify both radical generation and data analysis, FPOP will become an increasingly attractive option for biotherapeutic discovery, development, and biosimilar characterization workflows.
Conflict of Interest Disclosure
J.S.S. discloses a significant financial interest in GenNext Technologies, Inc., an early-stage company commercializing technologies for protein higher order structure analysis.
References
- Kiselar J, Chance MR. High-Resolution Hydroxyl Radical Protein Footprinting: Biophysics Tool for Drug Discovery. Annu Rev Biophys. 2018;47:315-333.
- Zhang B, Cheng M, Rempel D, Gross ML. Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods (San Diego, Calif. 2018;144:94-103.
- Li X, Grant OC, Ito K, et al. Structural Analysis of the Glycosylated Intact HIV-1 gp120-b12 Antibody Complex Using Hydroxyl Radical Protein Footprinting. Biochemistry. 2017;56(7):957-970.
- Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16(12):2057-2063.
- Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77(14):4549-4555.
- Buxton GV, Greenstock CL, Helman WP, Ross AB. Critical-Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen-Atoms and Hydroxyl Radicals (.OH/.O-) in Aqueous-Solution. J Phys Chem Ref Data. 1988;17(2):513-886.
- Sharp JS, Tomer KB. Effects of anion proximity in peptide primary sequence on the rate and mechanism of leucine oxidation. Anal Chem. 2006;78(14):4885-4893.
- Xie B, Sood A, Woods RJ, Sharp JS. Quantitative Protein Topography Measurements by High Resolution Hydroxyl Radical Protein Footprinting Enable Accurate Molecular Model Selection. Sci Rep. 2017;7(1):4552.
- Chance MR. Unfolding of apomyoglobin examined by synchrotron foot printing. Biochem Biophys Res Commun. 2001;287(3):614-621.
- Kaur P, Kiselar J, Yang S, Chance MR. Quantitative protein topography analysis and high resolution structure prediction using hydroxyl radical labeling and tandem-ion mass spectrometry (MS). Mol Cell Proteomics. 2015;14(4):1159-1168.
- Sharp JS, Becker JM, Hettich RL. Analysis of protein solvent accessible surfaces by photochemical oxidation and mass spectrometry. Anal Chem. 2004;76(3):672-683.
- Guan JQ, Chance MR. Structural proteomics of macromolecular assemblies using oxidative footprinting and mass spectrometry. Trends in biochemical sciences. 2005;30(10):583-592.
- Gau BC, Sharp JS, Rempel DL, Gross ML. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal Chem. 2009;81(16):6563-6571.
- Watson C, Janik I, Zhuang T, Charvatova O, Woods RJ, Sharp JS. Pulsed electron beam water radiolysis for submicrosecond hydroxyl radical protein footprinting. Anal Chem. 2009;81(7):2496-2505.
- Vahidi S, Konermann L. Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds. J Am Soc Mass Spectrom. 2016;27(7):1156-1164.
- Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Anal Chem. 2005;77(10):3029-3037.
- Saladino J, Liu M, Live D, Sharp JS. Aliphatic peptidyl hydroperoxides as a source of secondary oxidation in hydroxyl radical protein footprinting. J Am Soc Mass Spectrom. 2009;20(6):1123-1126.
- Vahidi S, Stocks BB, Liaghati-Mobarhan Y, Konermann L. Submillisecond protein folding events monitored by rapid mixing and mass spectrometry-based oxidative labeling. Anal Chem. 2013;85(18):8618-8625.
- Zhang Y, Rempel DL, Zhang H, Gross ML. An improved fast photochemical oxidation of proteins (FPOP) platform for protein therapeutics. J Am Soc Mass Spectrom. 2015;26(3):526-529.
- Li Z, Moniz H, Wang S, et al. High structural resolution hydroxyl radical protein footprinting reveals an extended robo1-heparin binding interface. J Biol Chem. 2015;290(17):10729-10740.
- Niu B, Zhang H, Giblin D, Rempel DL, Gross ML. Dosimetry determines the initial OH radical concentration in fast photochemical oxidation of proteins (FPOP). J Am Soc Mass Spectrom. 2015;26(5):843-846.
- Xie B, Sharp JS. Hydroxyl Radical Dosimetry for High Flux Hydroxyl Radical Protein Footprinting Applications Using a Simple Optical Detection Method. Anal Chem. 2015;87(21):10719-10723.
- Riaz M, Misra SK, Sharp JS. Towards high-throughput fast photochemical oxidation of proteins: Quantifying exposure in high fluence microtiter plate photolysis. Anal Biochem. 2018;561-562:32-36.
- Sharp JS, Misra SK, Persoff JJ, Egan RW, Weinberger SR. Real Time Normalization of Fast Photochemical Oxidation of Proteins Experiments by Inline Adenine Radical Dosimetry. Anal Chem. 2018;90(21):12625-12630.
- Garcia NK, Deperalta G, Wecksler AT. Current Trends in Biotherapeutic Higher Order Structure Characterization by Irreversible Covalent Footprinting Mass Spectrometry. Protein Pept Lett. 2019;26(1):35-43.
- Abolhasani Khaje N, Mobley CK, Misra SK, et al. Variation in FPOP Measurements Is Primarily Caused by Poor Peptide Signal Intensity. J Am Soc Mass Spectrom. 2018;29(9):1901-1907.
- Hambly DM, Gross ML. Cold chemical oxidation of proteins. Anal Chem. 2009;81(17):7235-7242.
- Watson C, Sharp JS. Conformational analysis of therapeutic proteins by hydroxyl radical protein footprinting. AAPS J. 2012;14(2):206-217.
- Lin M, Krawitz D, Callahan MD, Deperalta G, Wecksler AT. Characterization of ELISA Antibody-Antigen Interaction using Footprinting-Mass Spectrometry and Negative Staining Transmission Electron Microscopy. J Am Soc Mass Spectrom. 2018;29(5):961-971.
- Zhu Y, Guo T, Park JE, et al. Elucidating in vivo structural dynamics in integral membrane protein by hydroxyl radical footprinting. Mol Cell Proteomics. 2009;8(8):1999-2010.