Analytical Development and Commercialization
Analytical Development and Commercialization
Analytical Development and Commercialization
Analytical Development and Commercialization
Analytical Sciences, Pharmaceutical Sciences and Clinical Supply
Analytical Sciences, Pharmaceutical Sciences and Clinical Supply
Content uniformity testing of solid dosage forms is a critical test in almost all aspects of the drug development process. In both early- and late-phase development, uniformity testing meets a regulatory requirement for clinical release as directed by agencies globally. In scale-up efforts and design of experiment (DOE) tests in formulation development, it aids in the understanding of the effects of variation of parameters involved in solid dose formulation, including excipient compositions, blend rates, drying times, and tablet compression forces. As an increasing number of variables are examined in such testing, the number of individual dosage form units which need to be examined in chemical assays can become considerably large. Additionally, the manpower and reagents needed to carry out this testing become resource intensive as the amount of testing expands.
Employing traditional “out of the box” automation technologies in content uniformity sample preparations is an effective approach in the analyses of a large amount of individual tablets. However, such techniques suffer from the fact that they typically prepare samples serially rather than simultaneously, and that relatively large amounts of organic diluents are required for API extraction.
A novel and more environmentally friendly approach to content uniformity sample preparations was explored using an automated dissolution apparatus rather than traditional automated extraction platforms. With this approach, up to 6 individual units can be tested simultaneously in entirely aqueous diluents, often obtaining full API extraction in as little as 30 minutes. Proof of concept for this approach was demonstrated in multiple aqueous diluents for a test project and was then successfully employed in content uniformity testing of a large number of tablets for a program with accelerated timelines.
Background
Content Uniformity testing is an analysis required by worldwide agencies for release of a drug product batch for use in clinical studies. Stage I testing requires that a minimum of 10 individual dosage forms be examined for uniformity although this number can increase to a total of 30 units if Stage II testing is required. Evaluation of drug product in individual dosage forms is not limited to compendial release testing and may become crucial during scale-up efforts as a New Chemical Entity (NCE) prepares for commercial launch. Prior to New Drug Application (NDA) submission a variety of processing parameters may be examined in support of scale-up, including effect of compression force, blending times and speeds, and excipient composition. DOE Analyses are typically employed at this point of development, and to satisfy the test matrix, the number of samples to be formulated and tested can become exponentially large.
Rationale
Multiple reasons exist for exploring a novel, rapid, and resourcefriendly automated technique for preparation of content uniformity samples. First, existing automated extraction platforms available in the industry typically prepare samples serially or in a semi-serial method which results in a lengthy preparation process if multiple samples need to be tested. While these platforms can be employed effectively in use during non-work hours such as in overnight or weekend runs, operation during work hours is slow and inefficient, particularly if results are needed within the same day. Secondly, preparation of content uniformity samples is typically carried out in costly organic or hybrid aqueous/organic diluents which may be required in relatively large volumes for each sample preparation (typically 50 to 500 mL). As the number of samples increases (as in a DOE analysis), cost for reagents needed to carry out a test can become considerable. Also of note: costs for disposal of organic diluents are high and specialized as compared to disposal of wholly aqueous diluents. Preparation of samples in an entirely aqueous diluent, if possible, lowers the cost of analysis considerably. Lastly, the conventional content uniformity test will require some form of manpower for sample preparation and HPLC or UV analysis for the generation of results. If the content uniformity test is fully automated, the analyst is not needed at the bench and can focus efforts on more value-added activities as samples are prepared and analyzed on the automated platform.
A novel approach to preparation of content uniformity samples was explored using an automated dissolution apparatus. With this approach, individual dosage forms are added to each of 6 dissolution vessels and are agitated with paddles (USP apparatus II) until the dosage form is fully dissolved and the drug contained within is fully extracted into the media. This approach has the advantage that samples can be prepared at the same time rather than serially as with conventional extraction platforms. Additionally, diluents or media used in dissolution testing are totally aqueous in nature, which avoids the cost of expensive organic diluents and waste disposal methods. Lastly, automated dissolution typically requires little user interface for operation, and can be coupled with HPLC to effect a fully automated analysis. See Table 1 for a summary of advantages of the approach.
Table 1. Advantages of the Content Uniformity Test by Automated Dissolution

Cost Savings and Environmental Impact
Diluents employed in traditional content uniformity sample preparations are generally high in organic content, and can typically constitute anywhere from about 25% to 100% of the diluent composition. At costs of about $100 per liter of solvent (as in the case of acetonitrile, a highly used organic solvent in sample preparations), the costs of employing organic diluents in uniformity preparations can become exponentially large, particularly when expansive DOE studies are performed. By contrast, the content uniformity sample preparation by automated dissolution technique employs entirely aqueous diluents and avoids much of the high costs of diluent preparation and disposal of organic solvents. With these solvents, only relatively small amounts of buffer salts are added to water in order to prepare each, resulting in a near-negligible cost of materials required. In some cases, however, insoluble drugs may require the addition of surfactants such as sodium lauryl sulfate to achieve sink conditions. While costs of surfactants are similar to those of organic diluents on a per gram or per liter basis, these are generally used only in small amounts in media preparations (generally 0.5% to 3%) and so impact of use on cost is low as compared to that of organic solvents. Another consideration is that waste disposal of totally aqueous diluents is substantially less than that of organic solvents and the cost savings of such disposal is commensurate with the number of samples tested.
Experimental Design and Proof of Concept
A DOE analysis using a test compound that had been studied extensively by conventional analytical methodologies was selected for a proof of concept study for this novel automated approach to content uniformity testing. The DOE was constructed around the conventional non-automated dissolution method to evaluate the effects of ionic strength, pH, surfactant concentration, and surfactant type (ionic vs. non-ionic) for various dissolution medias that could be employed in testing. Ultimately, the DOE would determine dissolution test parameters that would result in the fastest dissolution profile possible so that the content uniformity sample preparations, when employed on automation, would be as short as possible. Testing employed the use of USP paddles (Apparatus II) at 250 RPM in order to achieve maximum agitation in the dissolution vessel, and samples were drawn at 15, 30, and 60 minutes to determine when full drug extraction occurred. See Table 2 for DOE plan and conditions used in analysis.
Table 2. DOE design employed in Proof of Concept Study. Paddle speed in testing was held constant at 250 RPM with samples drawn at 15, 30, and 60 minutes.

Dissolution profiles using the conditions prescribed by the DOE were essentially the same for all conditions studied, and full drug release was obtained between 30 and 60 minutes at 250 RPM in all cases. If the automated dissolution approach was employed in actual testing for this project, any media examined by the DOE could be used with agitation at 250 RPM and sample removal at 60 minutes successfully.
Thus, proof of concept for this approach was effectively demonstrated.
Equivalency and Employment in Testing
Table 3. Equivalency determination for two potencies of a compound to be studied in a process DOE. In both cases, the equivalency criteria of <2.5% difference in automated and manual methodologies was met.
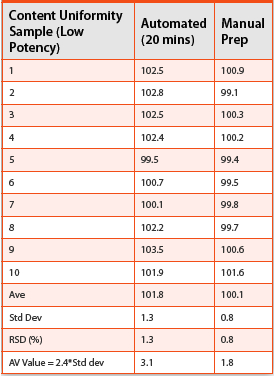
Following establishment of proof of concept, this novel automated approach was employed in analysis of samples in a process DOE for a project with accelerated timelines that routinely required rapid turnaround of data for any analysis performed. Before employment in actual testing, equivalency between the conventional manual methodologies and the automated approach was demonstrated for 2 potencies which would be tested in the DOE. Additionally, a carryover study was performed on the automation platform after analysis of the highest potency in order to demonstrate that the system was properly cleaning itself between testing of each batch of samples.
In order to establish equivalency, 10 individual samples of the same batch were tested by both automated and manual approaches for each potency and the mean and % RSD of each set were compared. See Table 3 for a summary of equivalency testing. Equivalency criteria of <2.5% difference in automated and manual methodology was used in comparison and was met for both potencies.
Following establishment of equivalency, content uniformity by automated dissolution was employed in a process DOE requiring sample preparation for a relatively large bolus of samples. In a time of approximately 2 days, a total of 84 individual sample preparations were carried out on the automated dissolution apparatus using entirely aqueous diluent.
 Figure 1. A comparison of Time required for manual uniformity sample preparations as compared to automated dissolution preparations.
Figure 1. A comparison of Time required for manual uniformity sample preparations as compared to automated dissolution preparations.Issues with Approach and SOP
This approach to content uniformity sample preparation is novel, resource-friendly, efficient, and amenable to any automated dissolution device. However, certain aspects of the testing must be considered before use, particularly for samples tested in a GMP environment. First, SOP governing equivalency criteria between the automated and manual sample preparations likely are non-existent as this approach is novel to the industry. While formulation of SOP is not critical for testing of samples of a non-GMP nature, an existing and approved SOP would need to be formulated before testing of samples for GMP purposes, such as in clinical release or in IND stability studies.
Additionally, chemical stability of a drug product in an entirely aqueous media versus that in an organic or mixed aqueous/organic solvent should be considered. This phenomenon would need to be examined on a case-by-case basis for each drug product considered for testing and may be critical if an API is only stable in an aqueous media for 24 hours or less.
Lastly, accuracy of each sample preparation will be proportional to the accuracy of volume of media delivered to each vessel by the automated dissolution device. In most cases, equipment vendors will require regular calibration of sample pumps or syringes which deliver media to each vessel. Some automated units also can perform gravimetric determinations of media delivered to each vessel so the exact volume in vessel can be derived from the mass of media. However, additional daily verifications may be required depending on quality restrictions at individual sites. This consideration is critical if samples to be tested on the device are GMP in nature.
Future Work
Automated dissolution equipment is efficient, easy to operate, and can automate most or all of the dissolution analysis. In addition to carrying out the steps of a typical dissolution test—such as heating of media, sample filtration, and cleaning of vessels— most systems have the ability to couple with HPLC or UV systems for full automation of the analysis. Content Uniformity testing by Dissolution on an automated system coupled with such back-end analytics potentially offers the fastest turnaround of data possible for this type of analysis.
Conclusion
Content Uniformity testing, while being a critical test in almost any aspect of dosage form development, is a labor intensive analysis that requires a large amount of manpower and resources for its completion. Traditional automated extraction platforms exist that can be employed to aid in this testing. However, these techniques suffer from the fact that they typically prepare samples serially and require large amounts of costly organic solvents in each sample preparation.
Analysis of individual tablets using automated dissolution has shown to be an effective alternative to traditional sample preparation methods. This approach can test multiple dosage units simultaneously and can employ entirely aqueous extraction medias for both manpower and resource savings. Following a demonstration of proof of concept, this approach was validated for use in a project with accelerated timelines and was employed successfully in a process DOE requiring analysis of a large bolus of samples. Future work will aim toward coupling the dissolution apparatus with in-line HPLC for full automation of the process. Additionally, SOPs will require modification of content to allow use of this approach.