Abstract
The discovery of RNA interference (RNAi) as a mechanism to selectively silence messenger RNA (mRNA) expression holds potential for revolutionizing biomedical research and drug development. The use of small interfering RNA (siRNA) as a target validation tool and therapy has become a major focus of industrial and academic laboratories. One of the primary challenges in realizing the full potential of siRNA therapeutics is the efficient systemic delivery of siRNA to the target cells. Liposome-based formulations in lipid nanoparticles (LNPs) have become the most promising and widely used strategy for in vivo delivery of siRNA. The complex nature of liposomal formulations containing duplex siRNA oligonucleotides and multiple functional lipid excipients presents a great challenge to the successful physicochemical evaluation of the siRNA LNPs. Ultra-high performance liquid chromatography (UHPLC) plays a critical role in characterizing the chemical properties of the LNP formulations. The high-throughput and superior separation efficiency offered by UHPLC enables the rapid and reliable determination of RNA, lipids, and related compounds in a complex formulation matrix. In this review article, an overview of the application of UHPLC for the chemical characterization of siRNA LNP formulations will be provided. Strategies for utilizing UHPLC potency and stability-indicating methods for siRNA oligonucleotides and lipids analyses will be described and future prospects will also be discussed.
Introduction
Discovered by Fire and Mello in 1998 [1], RNA interference (RNAi) is an endogenous process in which the translation of specific target messenger RNA (mRNA) in a cell is silenced in the presence of small complimentary RNA sequences which bind the mRNA and activate the RNA silencing complex (RISC). Targeting of exogenous RNA sequences to cell cytosol also activates RISC and presents substantial opportunity as a therapeutic and target validation tool. Industrial and academic communities have quickly capitalized on the discovery and adopted RNAi as a powerful target validation tool and a promising new therapeutic modality [2, 3]. As the primary focus of research into the utilization of RNAi as a therapy, siRNA (small interfering RNA) is a chemically synthesized RNA duplex and is generally delivered by nonviral delivery systems [4, 5]. Because of its poor pharmacological properties including serum instability, urine excretion, and inefficient cell delivery, effective and safe systemic delivery of siRNA to the target tissues has become the major hurdle in realizing the full potential of RNAi as drug therapeutics. Among various approaches to overcome the challenges, cationic liposome-based formulations in lipid nanoparticles (LNPs) represents the most promising and widely used strategy for in vivo delivery of siRNA [6,7]. A distinct advantage of the cationic LNPs is their ability to greatly improve the pharmacokinetic properties of synthetic oligonucleotides administered systemically. Efficient entrapment of the highly hydrophilic and negatively charged duplex siRNA oligonucleotides inside positively charged nano-particles protects siRNA from enzymatic degradation leading to significantly improved biodistribution and intracellular delivery [8, 9].
In addition to duplex siRNA oligonucleotides as the active pharmaceutical ingredient, an siRNA LNP formulation generally consists of multiple lipids that play an important role in defining the LNP functions [Figure 1]: a cationic lipid (CL) to enable the encapsulation of oligonucleotides and the binding of LNP to anionic cell surface molecules; a “stealthy” pegylated-lipid conjugate (PEG-lipid) to control LNP particle size during formulation and prolong its systemic circulation time; and neutral “helper” lipids including a phospholipid to enhance the fusogenicity and cholesterol to minimize thermotropic phase transitions. A thorough physicochemical evaluation of LNP is essential for the siRNA liposomal formulation development and optimization. Furthermore, as RNAi therapies advance from preclinical to clinical development, demand for rigor in CMC definition of the oligonucleotide and delivery components rises. To assure product quality and process capability, more advanced analytical methodology for the analysis of siRNA LNPs is evolving [10].
In order to accomplish the full characterization of these complex formulations, multiple analytical techniques have typically been employed including chemical analysis, particle size analysis, and morphological analysis [8]. Rapid determination of RNA and lipid contents enables efficient formulation development, process optimization and manufacturing control. More important, reliable stability-indicating methods are required to ensure the purity and stability of the LNP products used in the preclinical safety and clinical studies. High performance liquid chromatography (HPLC) is the primary tool for the chemical characterization of LNPs. The complex nature of liposomal formulations containing siRNA duplexes and multiple functional lipid excipeints presents a significant chromatographic challenge. Both siRNA oligonucleotides and lipids can degrade by various pathways including hydrolysis, oxidation, and desulfurization, potentially generating numerous impurities and degradates in the LNP formulations. The need to detect and quantitate RNA and lipids related degradation products has placed great demands on the chromatographic technology for the analysis of siRNA LNP. Ultra-high performance liquid chromatography (UHPLC), which offers significant advantages over HPLC in terms of sample throughput and resolving power, is an excellent alternative for the analysis of siRNA LNPs. In this review article, an overview of the application of UHPLC for the chemical characterization of siRNA LNP formulations will be provided. Strategies for developing UHPLC potency assays and stability-indicating methods for RNA and lipids analyses will be described and future prospects will also be discussed.
UHPLC in Pharmaceutical Analysis
Since the first commercial ultra-high pressure liquid chromatograph system was introduced in 2004, UHPLC has gained increasingly popularity and wide acceptance across many industries. Although it has yet to replace conventional HPLC systems as the work horse in the pharmaceutical laboratories, UHPLC has been implemented in many segments of drug discovery and development. UHPLC technology promises significantly faster separations in addition to increased chromatographic resolution and enhanced “mass” sensitivity. Specially plumbed UHPLC hardware offers low overall system dispersion for improved separation efficiency, small delay volume enabling rapid column re-equilibration for gradient analysis, fast detector response for narrowly eluting peaks. These benefits of UHPLC make it well suited for fast and ultrafast potency assays without significant loss of separation efficiency [11]. Normally, 4-10 folds of increase in speed can be obtained using UHPLC. Consequently, high laboratory productivity desired to speed up the drug discovery and pre-clinical development can be realized with UHPLC, ultimately lowering the ever-increasing drug development costs in today’s pharmaceutical industry. Besides its capability to achieve fast and ultrafast separation, another significant advantage of UHPLC is its capability of solving the most challenging separation tasks in pharmaceutical analysis. By offering much higher operating pressure limits (up to 1200-bar) compared to standard HPLC (400-bar), UHPLC allows the use of sub-2 μm (STM) particle columns of lengths up to 150 mm to achieve very high separation efficiencies and increased sensitivity, resulting in considerable benefits in high resolution LC analysis since the STM columns provides much higher theoretical plate counts than the conventional HPLC columns packed with 3-5 μm particles. Moreover, when combined with high column temperature and column coupling, UHPLC can achieve ultra-high resolution separation for the analysis of very complex mixtures, where the high separation efficiency often far outweighs the selectivity optimization due to the presence of multiple critical pairs needed to be resolved chromatographically [11]. For example, more than 100,000 plates were generated for heptanophenone in under 12 min with three linked C18, 2.1 x 150mm, 1.7μm columns operated at 90 °C and 14,100 psi [11]. The unique characteristics of UHPLC make it especially suited for the analysis of siRNA LNP products as demonstrated in the following sections.
Analysis of siRNA Oligoneucleotides by UHPLC
siRNA is a class of double-stranded RNA (dsRNA) molecules, 21-23 nucleotides (nt) in length, with 2-nt 3’ overhangs on both passenger strand (PS) and guide strand (GS) [Figure 1]. Among many analytical techniques utilized for the analysis of oligonucleotides including radiolabel tracer methods, ELISA methods, gel electrophoresis-based methods, and HPLC, strong anion-exchange LC (SAX-LC) and ion pairing reserved phase LC (IP-RP LC) with UV-visible detection at 260- nm have been the standard techniques for the analysis of therapeutic siRNAs as a result of HPLC’s dominant presence in pharmaceutical labs. SAX-HPLC is a traditional technique for nucleic acid analysis and most commonly used for the identification and potency determination of siRNA duplex in LNPs against placebo siRNA sequences and/or undesirable non-hybridized single-stranded RNAs (ssRNA) which are often associated with a decrease in therapeutic potency. SAX analysis is normally performed using non-denaturing mobile phases and low separation temperature (≤ 40 °C) to maintain the integrity of non-covalent siRNA duplex throughout the analysis. Although the development of small particle SAX-LC columns has lagged significantly behind the ones for RP-LC, the first commercial STM SAX columns has been introduced very recently [12]. A preliminary evaluation of the column on the UHPLC in our lab has produced promising results, indicating a potential significant improvement in resolution and speed compared to the commonly used SAX HPLC columns as shown in Figure 2. Three of four siRNAs with very similar structures were seen co-eluting on a 25-cm, 8-μm SAX-HPLC column in a 20-min separation while a rapid separation with much improved resolution was achieved with a 5-cm STM column on UHPLC in less than 2 minutes.
Compared with SAX-LC for oligonucleotide analysis, IP-RP LC offers higher separation efficiency and compatibility with mass spectrometry (MS), and has increasingly become the method of choice for impurity profiling of siRNAs. Conventional IP-RP separation methods for oligonucleotides are based on the use of triethylammonium acetate (TEAA) containing mobile phases at pH 7.0. Mass transfer in the stationary phase was found to be a major factor contributing to peak broadening on porous C18 columns for analysis of oligonucleotides [13]. Small particle size, elevated temperature and a relatively slow flow-rate have been recommended to maximize separation efficiency. Consequently, STM columns combined with UHPLC would provide significant advantages over IR-RP HPLC. Benefits of IP-RP UHPLC for siRNA analysis including improved resolution and productivity have been demonstrated with approximately four-fold increase of sample throughput compared to traditional IR-RP HPLC methods [14].
Major sources of siRNA related impurities in LNPs are often the result of multiple degradation pathways such as hydrolysis of the phosphoester bond and the N-Glycosidic bond, cleavage of bases and strand scission due to oxidation, leading to truncated oligonucleotides [(N-x) impurities] [15]. Additionally, chemical modifications to siRNA backbone, sugars, and/or bases are usually made to enhance the performance of siRNA, including increased serum stability, decreased off -target effects, and reduction of undesired immune stimulation. These modifications have made analysis and characterization of siRNAs more challenging than their native counterparts. For example, integration of phosphorothioate (P-S) backbone linkages by the substitution a sulfur atom for one of the non-bridging oxygen atoms in each phosphodiester bond provides exonuclease resistance (Figure 3). However, P-S backbone has been found to be susceptible to oxidation and can undergo desulfurization to generate the phosphodiester (PO) form which undermines siRNA drug in vivo stability. Therefore, it is critical to have a stability-indicating method which is specific against the P-O degradates as well as the (N-x) impurities. Furthermore, sulfurization of oligonucleotide backbone creates multiple new diastereometers, causing substantial peak broadening in IP-RP HPLC analysis [16]. Chromatographic resolution of P-S diastereomers can be difficult or impossible to resolve by the conventional IP-RP HPLC method using TEAA ion-pairing system [16].
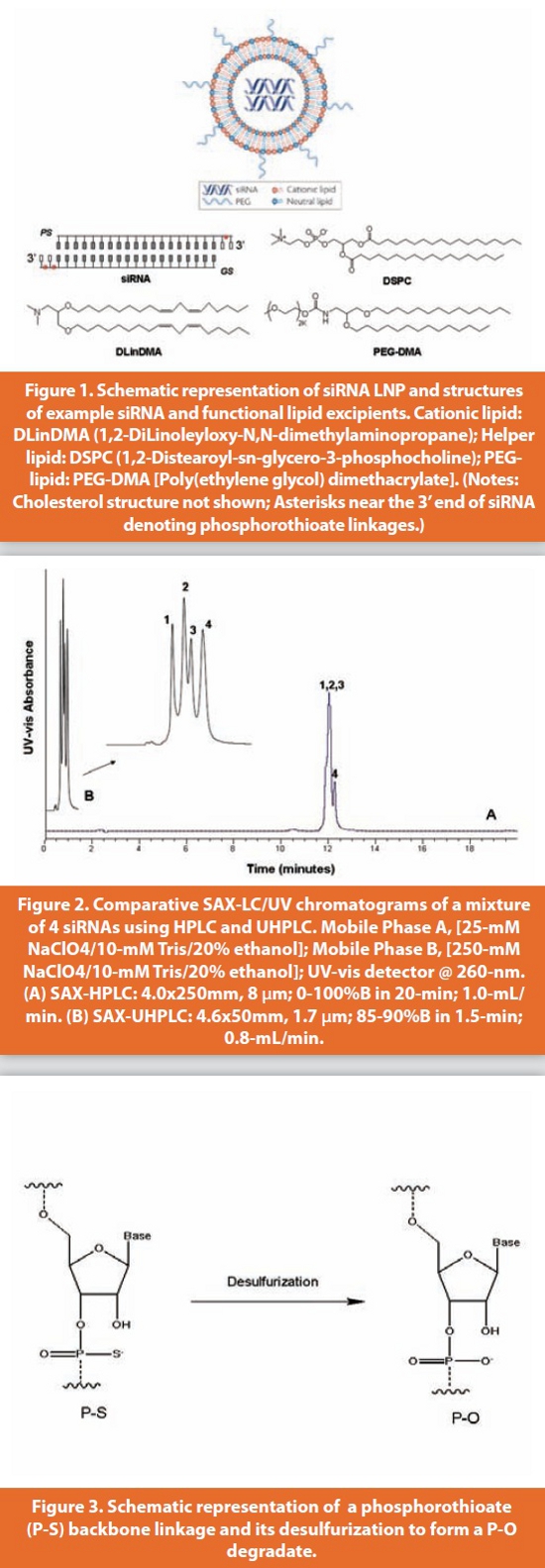
Direct impurity profiling of siRNA duplex is particularly challenging because of the small differences in size, charge and hydrophobicity between the full length duplex, ssRNAs, and impurities, which often co-elute. Therefore, IP-RP LC analysis of impurities in siRNAs is typically performed at elevated column temperature to fully denature the duplex and generate two single-stranded counterparts. Figure 4 illustrates the IP-RP analysis of a P-S siRNA on UHPLC utilizing in-situ denaturation of the siRNA duplex at 80 °C. Resolution of two GS diastereomers and four PS diastereomers were achieved on a 15-cm STM column within 20 mins. To further take the advantages of very high operating pressure limit offered by UHPLC, column-coupling (40- cm) was employed to fully resolve the ssRNA peaks from their (N-x) impurities. The improved resolution achieved with a longer column provided for increased method robustness to ensure the detection and reliable quantitation of potential new impurities and degradates.
The IP-RP UHPLC method with TEAA IP buffer is suitable for the impurity profiling of siRNA products as a routine process control and product release. However, monitoring degradation profiling in the stability testing can be challenging due to the presence of numerous diastereomer peaks for both full length RNA and (N-x) impurities. IP aqueous buffers composed of triethlyamine (TEA) and hexafluoropropanol (HFIP), pH 7.9, and methanol have been found to tend to suppress the diastereomeric resolution, and therefore, are more suitable for stability testing of P-S oligonucleotides [16-17]. In addition, the TEA/HFIP/methanol based mobile phases do not cause ion suppression and are more appropriate for LC/MS analysis of unknown degradation products. Figure 5 compares the IP-RP separation achieved on HPLC and UHPLC with TEA/HFIP/methanol system under the fully-denaturing condition (80 °C). The figure illustrates that the UHPLC method provides significantly improved resolution and sensitivity for detection of potential degradation products including P-O and (N-x) degradates compared with the HPLC method. Separation performance of UHPLC makes it an ideal chromatographic technique for the analysis of therapeutic P-S oligoneucleotides. It should be pointed out that chromatography techniques alone, even with extremely high separation efficiency achieved with UHPLC, cannot solve all separation problems and unequivocally identify degradation products. LC coupled with electrospray ionization mass spectrometry (ESI-MS) has increasingly being used as an important characterization tool for oligonucleotides, enabling identification of process-related impurities and degradation products [16-18]. UHPLC coupled with MS to deliver superior LC performance for optimal LC/MS characterization of siRNAs, is especially suited for the quantitation of P-O degradates. The mass difference of 16 Da between all P-S oligonucleotides and their monodiester forms can be readily detected using deconvoluted ESI-MS signal [16].
Analysis of Functional lipid Excipients by UHPLC
Unlike the commonly used excipients in the conventional oral and parenteral formulations, the lipid composition as well as siRNA/total lipids ratio are the critical parameters to define the bio-performance of the siRNA LNPs. Therefore, development of reliable analytical methods for thoroughly unraveling the composition of functional lipid excipients and related impurity profile is essential in the development of LNP formulations.
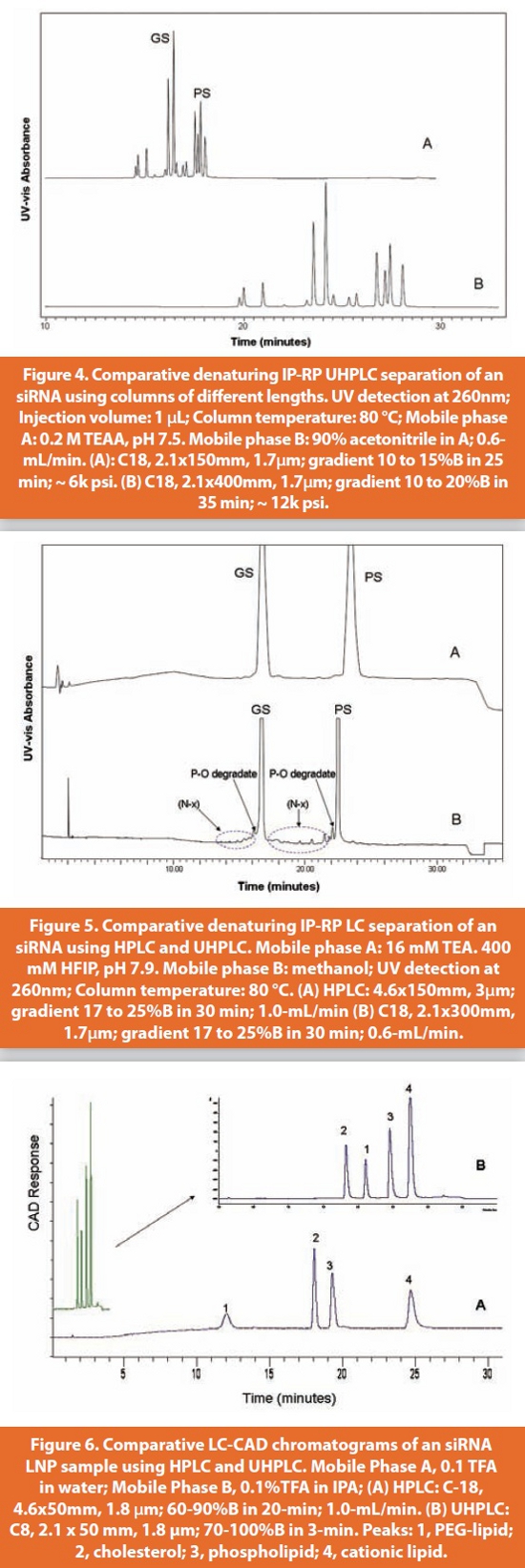
The diversity in structural features along with the distinct hydrophobic and hydrophilic elements makes lipids one of most difficult group of substances to analyze. PEG-lipids can be particularly challenging due to their polydispersity as a result of molecular-weight distribution. Compared to RNA, one additional challenge for lipids analysis is that most of lipids lack chromophores that might facilitate the use of the standard UV-vis detector for LC. The situation has made the use of a universal HPLC detection technique necessary such as the ones based on aerosol particles produced via nebulization including evaporative light scattering detector (ELSD) and charged aerosol detector (CAD). Unlike ELSD which correlates the amount of light scattered with analyte concentration, CAD utilizes a measurement of charge that is carried by the analyte to an electrometer for correlation [19]. Compared to the ELSD for the lipid analysis, the CAD has been demonstrated to offer a more uniform response curve over a wider range of concentrations (over 4 orders of magnitude) with better accuracy, precision and lower limits of detections (typically an order of magnitude better than ELSD) [20-22], which are the critical attributes for HPLC methods to quantitate low levels of impurities in pharmaceutical products. In addition, CAD has been found to be more user-friendly since it does not require any optimization or operating parameters to be set, except for the output range, while the ELSD requires some optimization in nitrogen pressure, drift tube temperature, and nebulizer temperature in order to attain maximum sensitivity [23]. Since the introduction of the fi rst commercial CAD in 2004, it has been widely used throughout pharmaceutical and biopharmaceutical R&D. An HPLC-CAD method was recently developed and found suitable for the detection of lipids in liposomal formulations [24]. A comparison of HPLC and UHPLC separation for the analysis of lipid composition in an siRNA LNP product is shown in Figure 6. Compared to HPLC, a gain in analysis speed by factors of 10 is realized without sacrificing the separation efficiency. In addition, the much sharper peak shape obtained in the UHPLC chromatogram, especially for the PEG-lipid, makes the peak integration more robust, leading to improved accuracy in quantitation.
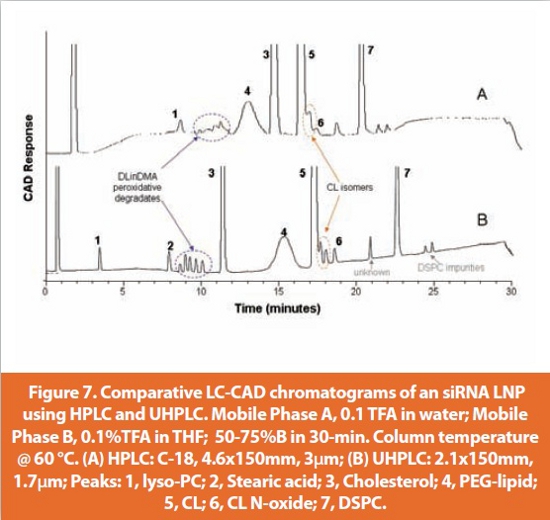
There are potentially several mechanisms by which the lipid components in an siRNA LNP formulation can undertake to generate degradates which will compromise the product stability and bioperformance. Free-radical-initiated lipid peroxidation is a potential route of degradation for the cationic lipid (CL) containing unsaturated linoeyl hydrocarbon chains such as DLinDMA (Figure 1) and can produce a variety of complex degradation products. CL with a tertiary amine head group may also undergo two-electron oxidation to form N-oxide. Hydrolysis is the primary degradation mechanism for estercontaining lipids such as DSPC (Figure 1), which will produce lysophosphatidylcholine (lyso-PC) and stearic acid, resulting in a lower pH of the formulation. It is essential that a stability-indicating method for lipids is specific against all of these potential degradation products in addition to the synthetic impurities of the lipids such as the CL isomers. Figure 7 shows an impurity separation under conventional HPLC conditions with a 3-μm particle, 4.6x150mm column, and the same separation performed via UHPLC using a 2.1x150 mm column with 1.7-μm particles. Exceptional resolution of all potential degradation products and synthetic impurities was obtained with the UHPLC method within 30 mins. Although no reduction in run time was achieved compared to the HPLC method, the high efficiency of the UHPLC separation allowed the increased resolution between the DLinDMA peroxidation degradates and stearic acid as well as closely eluting DLinDMA isomers and N-oxide peaks, which elute right after the parent peak.
Conclusion
Gene silencing by RNA interference has shown great promise in biomedical research and therapeutic applications. The development of safe and efficient in vivo systemic delivery systems remains as a primary hurdle for realizing the full potential of siRNA therapeutics and necessitates a thorough physicochemical characterization of siRNA LNPs. Ultra-high performance liquid chromatography plays a critical role in characterizing the chemical properties of the LNP formulations. In comparison to traditional HPLC, the high-throughput capability offered by UHPLC along with UV-vis and charged aerosol detectors enable the accurate and rapid potency determination of siRNA oligonucleotides and functional lipid excipients in the chromatographically challenging sample matrices, leading to greater overall laboratory throughput and efficiency. The superior resolving power of UHPLC provides for the reliable impurity profiling of numerous RNA and lipids related substances associated with the multiple degradation pathways, facilitating regulatory requirements compliance for the assurance of purity and stability of the siRNA products. UHPLC-MS is becoming a popular tool for siRNA analysis because of its capability to identify oligonucelotide impurities and degradates and to quantitate the analytes which are not or only partially chromatographically resolved. It seems likely that UHPLC-MS will play an increased role in routine quality control analysis of siRNA therapeutic products.
References
1. A. Fire, S. Xu, M. Montgomery, S. Kostas, S. Driver, C. Mello, Nature 391 (1998) 806-811.
2. L. Aagaard, J.J. Rossi, Advanced Drug Delivery Reviews 59 (2007) 75-86.
3. Q. Leng, M.C. Woodle, P.Y. Lu, A.J. Mixson, Drugs of the Future 34 (2009) 721-737.
4. L. Li, Y. Shen, Expert Opin. Biol. 9 (2009) 609-619.
5. A.R. De Fougerolles, Human Gene Therapy 19 (2008) 125-132.
6. Y.C. Tseng, S. Mozumdar, L. Huang, 61 (2009) 721-731.
7. S.Y. Wu, N.A.J. McMillan, The AAPS Journal, DOI: 10.1208/s12248-009-9140-1.
8. I. MacLachlan, Antisense Drug Technologies, 2nd Ed, Chapter 9, (2007) 237-270.
9. S.C. Semple, et al., Nature Biotechnology 28 (2010) 172-176.
10. V. Murugariah, W. Zedalis, G. Lavine, K. Charisse, M. Manoharan, Anal. Biochem. 401 (2010) 6167.
11. Z. Li, American Pharm. Rev. 11 (2008) 24, 26, 28-31.
12. Agilent Application Notes (2010) 5990-5195EN.
13. M. Gilar, K.J. Fountain, Y. Budman, U.D. Neue, K.R. Yardley, P.D. Rainville, R.J. Russell II, J.C. Gebler, J. Chromatogr. A 958 (2002) 167-182.
14. M. Gilar, Waters Application Notes (2007) 720002383EN.
15. D. Pogocki, C. Schoneich, J. Pharm. Sci. 89 (2000) 443-455.
16. M. Gilar, K.J. Fountain, Y. Budman, J.L. Holyoke, H. Davoud, J.C. Gerbler, Oligonucleotides 13 (2003) 229-243.
17. A. Apffel, J.A. Chakel, S. Fischer, K. Lichtenwalter, W.S. Hancock, Anal. Chem. 69 (1997) 1320-1325.
18. M.B. Beverly, Mass Spectrometry Rev. (2010) DOI 10.1002/mas.20260.
19. M. Plante, B. Bailey, I. Acworth, Methods in molecular biology, 579 (2009) 469-82.
20. L.M. Nair, J.O. Werling, J. Pharm. Biomed. Anal. 49 (2009) 95-99.
21. R.G. Ramos, D. Libong, M. Rakotomanga, K. Gaudin, P.M. Loiseau, P. Chaminade, J. Chromatogr. A 1209 (2008) 88-94.
22. C. Merle, C. Laugel, P. Chaminade, A. Baillet-Guffroy, J. Liq. Chromatogr. Rel. Technol. 33 (2010) 629-644.
23. N. Vervoort, D. Daemen, G. Torok, J. Chromatogr. A 1189 (2008) 92-100.
24. C. Schonherr, S. Touchene, G. Wilser, R. Peschka-Suss, G. Francese, J. Chromatogr. A 1216 (2009) 781-786.
Author Biographies
Dr. Zhong Li is currently a research fellow at RNAi Analytical Sciences, Pharmaceutical Science of Merck Research Laboratories at West Point, Pennsylvania. He received his B.S. in Inorganic Chemistry and M.S. in Analytical Chemistry from Shandong University, Jinan, China. He earned his Ph.D. in Analytical Chemistry in 1999 at the University of Alberta, Edmonton, Canada. Dr. Li’s main scientific interests lie in the areas of high-performance liquid chromatography (HPLC), gas chromatography (GC), high-throughput analysis, headspace sampling, mass spectrophotometry and their applications in pharmaceutical and biopharmaceutical analysis.
Mr. Joe Schariter is currently a research chemist at RNAi Analytical Sciences, Pharmaceutical Sciences of Merck Research laboratories at West Point, Pennsylvania. He received his B.S. in Biochemistry with a Minor in Economics from Lafayette College in Easton, Pennsylvania. He is currently earning his M.S. in chemistry from Lehigh University and his interests lie in chemical characterization of pharmaceutical and biopharmaceutical products using UHPLC, mass spectrometry, and gas chromatography.
Dr. Jingtao Zhang is a senior research biochemist at RNAi Analytical Sciences, Pharmaceutical Science of Merck Research Laboratories at West Point, Pennsylvania. His past and current roles include formulation, physical characterization, and analytical testing on lipid-based siRNA delivery vehicles to support the clinical development of siRNA therapeutics. Dr. Zhang received his Ph.D. in Chemical Engineering at the University of Wisconsin-Madison in 2007. He received his B.E. (1999) from Nanjing University of Chemical Technology, China and M.S. (2002) from Tsinghua University, China. His main research interests are in the area of siRNA and gene delivery, biomaterials and controlled release, nano-material formulation and characterization, biophysical and characterization techniques to enable well-characterized biopharmaceutical products.
Mr. Jared Davis is currently a staff chemist at RNAi Analytical Sciences, Pharmaceutical Science of Merck Research Laboratories at West Point, Pennsylvania. He graduated from the Pennsylvania State University in 2004 with a B.S. in Chemistry. Jared has been with the RNAi Analytical Sciences team since joining Merck in 2007. His work is heavily focused on developing characterization assays for nucleic acid lipid nanoparticles using high-performance liquid chromatography.
Dr. Anthony Leone is currently an Associate Director leading the RNAi Analytical Sciences group at Merck Research Laboratories in West Point, PA. He earned his BA in Chemistry from Franklin & Marshall College in 1998, and PhD in Chemistry from the University of North Carolina-Chapel Hill in 2003. He joined Merck in 2003 working in Pharmaceutical R&D and has been a key contributor and leader of multiple small and large molecule discovery and development programs. His research interests are centered on macromolecular and nanoparticle characterization.