Introduction
The primary task within drug discovery in the pharmaceutical industry is to bring forth compounds that have the best overall profile. This insures the highest chance for success within drug development where new chemical entities fail for reasons, such as toxicity, lack of efficacy and poor human pharmacokinetics. Additionally, it is critical to remove compounds that have no chance of success from the pipeline as early as possible in the drug discovery process so that money and resources may be allocated on “potential winners” and not wasted on the “losers.” We, therefore, take a two-pronged approach in drug discovery: i) find the best compounds and progress them as quickly as possible, and ii) “kill” the “losers” as quickly as possible. There are many “filters” or screens that determine the fate of a compound and constitute a go/no go decision in early drug discovery. One of the most critical “filters” is in vivo rat pharmacokinetics (PK) [1-3]. Clearly, if the requisite plasma concentrations for efficacy and drug safety are not achieved in rats, then, in most cases, the drug has no chance of success. It stands to reason that obtaining this data in as short a timeframe as possible is critical within drug discovery. There are various limiting factors within the development of such a paradigm, including resources, communication and instrumentation.
We have developed a novel and streamlined approach that avoids classical “cassette dosing” [4, 5] and its potential pitfalls. The Cassette-Accelerated Rapid Rat Screen (CARRS) [6] is a high throughput in-vivo screen that provides the earliest PK information on 72 new chemical entities (NCEs) per week as shown in Figure 1. 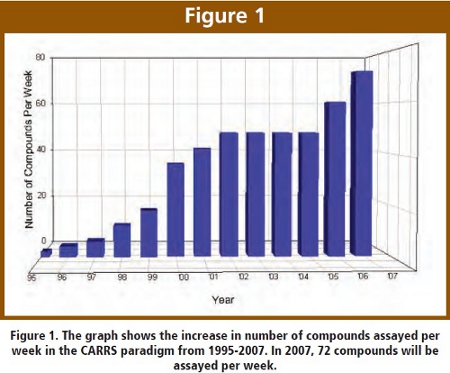 Six NCEs are selected per cassette for a total of 12 cassettes. Two rats are dosed with each NCE and plasma samples are collected at 0.5, 1, 2, 3, 4 and 6 hours post-dose. Tissue samples may also be harvested at the six hour time-point for specific programs. The total turnaround time for dosing, method development, analysis and report submission is two weeks. In a recent study, Mei et al. found good correlation between CARRS and full rat PK studies [7].
Six NCEs are selected per cassette for a total of 12 cassettes. Two rats are dosed with each NCE and plasma samples are collected at 0.5, 1, 2, 3, 4 and 6 hours post-dose. Tissue samples may also be harvested at the six hour time-point for specific programs. The total turnaround time for dosing, method development, analysis and report submission is two weeks. In a recent study, Mei et al. found good correlation between CARRS and full rat PK studies [7].
The primary tool for determining drug concentrations in biological matrices is high performance liquid chromatography combined with tandem mass spectrometry (LC-MS/MS). This technique provides a two dimensional approach that is both highly sensitive and selective for the analyte of interest. Limits of quantitation in the 1-10 ng/mL range and a dynamic range over three to five orders of magnitude are now routine. Instrumentation has become more compact, rugged and easy to use so that multiple systems are routinely found in many drug discovery and development metabolism and pharmacokinetic laboratories. Recent advances in HPLC column technology, combined with fast scanning mass spectrometers, has resulted in shorter cycle times and narrower peak widths.
We use LC-MS/MS to analyze biological samples within drug discovery. In addition to providing quantitative information on the parent drug, it is also useful to monitor for select metabolites. This serves two functions: to help chemists identify metabolic “hot spots” and help identify a new lead compound or prodrug.
We have previously explored the use of short HPLC columns and ultra fast gradients in order to decrease LC-MS/MS analytical run times [8]. Typical cycle times ranged from 85 seconds to two minutes and used 2 x 30 mm, 4 μm HPLC columns (Table 1). The major drawback with this approach is loss of separation resolution at higher linear velocities, as demonstrated by the van Deempter curve. Recently, advances in HPLC column technology have made small particle size columns (average particle size 1.7 – 2.1μm) readily available. These smaller particles dramatically increase column efficiency, which has a large impact on the speed of LC-MS/MS applications. The major drawback in using small particles is maintaining stable flow rates and the increase in column backpressure requiring the use of pumps with higher pressure ratings (15,000 psi). We have used these small particle columns in conjunction with high pressure pumps to create a true ultra fast chromatographic approach to pharmacokinetics and metabolism.
In order to fully appreciate the benefit of using small particle column packing material, one must examine the van Deempter equation, which is fundamental to chromatographic theory. According to the van Deempter equation:
H = A + B/μ + Cμ
where H is the column’s plate height and μ is the mobile phase flow rate. A, B and C are constants. A smaller plate height equals a greater separation resolution, which is vital for separating the analyte of interest from its metabolites and from endogenous material in the matrix. The A and C constants are dependent on the particle size and therefore a decrease in particle size will result in a decrease in H. However, as the flow rate increases, the C term becomes more important and results in a loss of resolution above a certain flow rate. The benefit of using 2 μm or smaller particles is that it allows a significant increase in flow rate before a loss of resolution. In the world of LC-MS/MS, this translates into the equivalent of having your cake and eating it, too. In a typical LCMS/ MS analysis, there is a trade-off between speed, resolution and sensitivity. With the advent of small particle HPLC columns, it is possible to attain all three without sacrificing anything [9].
HPLC vs. Ultra Fast HPLC
In order to have a true comparison between conventional HPLC and ultra fast HPLC using small particles, it is necessary to compare them head to head using the same compounds. For this study, we used a series of eleven commercially available compounds, shown in Figure 2. Our standard HPLC conditions are shown in Table 1. Mobile phase A is methanol/water (20:80), 0.010 M ammonium acetate and mobile phase B is methanol/water (90:10), 0.010 M ammonium acetate, 0.2 percent of ten percent acetic acid. Under our “old” HPLC conditions, typical cycle time of 2.5 minutes is easily achievable. The analyte of interest elutes at a retention time of 1.2 – 1.6 minutes with a run time of two minutes, then approximately 30 seconds is needed for the mass spectrometer to communicate with the autosampler in preparation for the next sample. In the CARRS paradigm, a cassette of plasma samples requires 108 injections, including standards, samples and blanks. Therefore, the total run time is 4.5 hours and increases to seven hours for a plasma and tissue cassette. This precludes same day turnaround and does not allow for the most efficient use of automated method development software. Ultra fast HPLC can achieve 1 – 1.5 minute cycle times, which allows for same day turnaround of a plasma and tissue cassette and the completion of four to seven cassettes in an overnight run. This allows for rapid turnaround of data in a highly efficient manner.
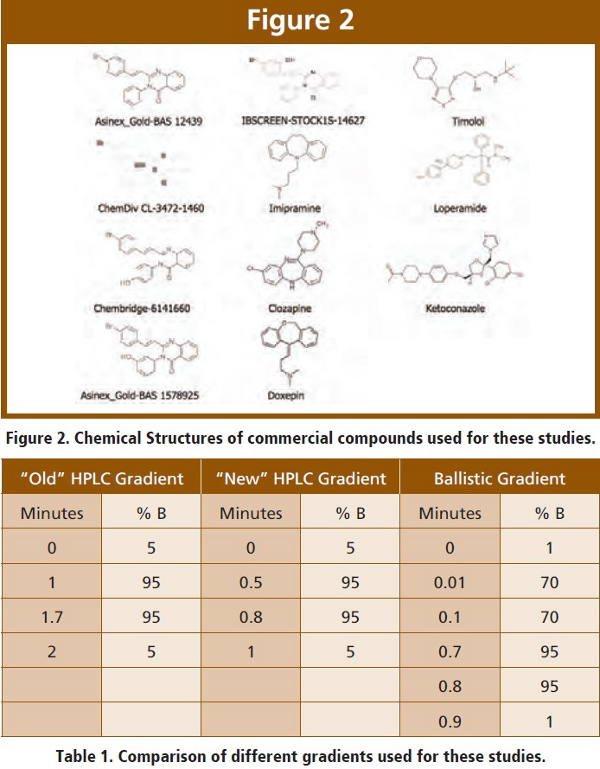
Figure 3 shows the comparison between a typical HPLC (2 x 30 mm, 4μm) run and ultra fast HPLC (2 x 30 mm, 1.7μm) for BAS 12439 and its hydroxy analog (CL-3472-1460). The ultra fast HPLC run used a gradient that ramps from 10 percent B to 100 percent B between 0.1 and 0.15 minutes. The flow rate was 0.9 mL/min resulting in a backpressure of approximately 14,000 psi. The HPLC run ramps from 10 percent B to 100 percent B between 0.2 and 0.5 minutes. A threefold improvement in speed and three-fold decrease in peak width is observed. This is a relatively straightforward example of what is possible with ultra fast HPLC. Similar data has been generated for many different compounds. 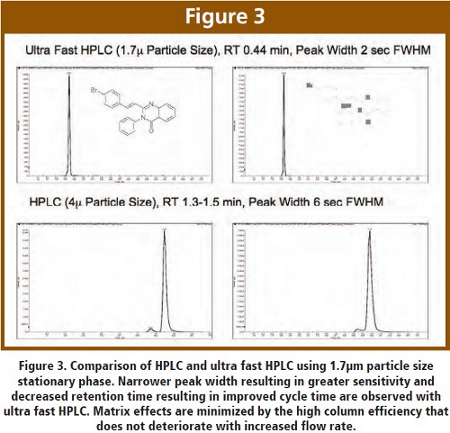 The key feature is the increase in speed while maintaining or improving sensitivity and quality. Figure 4 shows another example of ultra fast HPLC, using a different high pressure pump and a different column for imiparamine, doxepin, clozapine and timolol with retention times of 0.3-0.4 min and peak widths of 3-4 sec FWHM. The ballistic gradient represents what is possible for ultra fast HPLC. In practice, we use a less aggressive approach that yields retention times of 0.5-0.8 minutes. This is sufficient for same day analysis, as well as the analysis of seven 96 well plates in 24 hr. Also shown in Figure 4 is the corresponding CARRS standard curve (25, 250, 2500 ng/mL), which demonstrates good linearity across the region of interest. The low end of the standard curve is easily achieved with minimal method development. We employ generic gradients and automated method development for over 90 percent of our assays with little difficulty.
The key feature is the increase in speed while maintaining or improving sensitivity and quality. Figure 4 shows another example of ultra fast HPLC, using a different high pressure pump and a different column for imiparamine, doxepin, clozapine and timolol with retention times of 0.3-0.4 min and peak widths of 3-4 sec FWHM. The ballistic gradient represents what is possible for ultra fast HPLC. In practice, we use a less aggressive approach that yields retention times of 0.5-0.8 minutes. This is sufficient for same day analysis, as well as the analysis of seven 96 well plates in 24 hr. Also shown in Figure 4 is the corresponding CARRS standard curve (25, 250, 2500 ng/mL), which demonstrates good linearity across the region of interest. The low end of the standard curve is easily achieved with minimal method development. We employ generic gradients and automated method development for over 90 percent of our assays with little difficulty.
Table 2 shows a comparison of ultra fast, standard and ballistic HPLC for BAS 12439. The ultra fast HPLC utilized a gradient that ramped from 40 percent B to 100 percent B in 0.2 minutes, normal HPLC ramped from 10 percent B to 100 percent B in one minute and ballistic HPLC ramped from 10 percent B to 100 percent B in 0.1 minute. 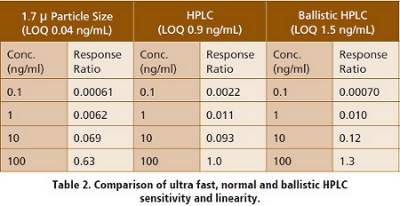 The LOQ is defined as 40 percent of the lowest usable standard or three times the back-calculated blank (standard “0”) value, whichever is greater. The results observed in Table 2 are typical for what is routinely observed. Linearity, dynamic range and sensitivity are all under ultra fast HPLC conditions.
The LOQ is defined as 40 percent of the lowest usable standard or three times the back-calculated blank (standard “0”) value, whichever is greater. The results observed in Table 2 are typical for what is routinely observed. Linearity, dynamic range and sensitivity are all under ultra fast HPLC conditions.
Ultra Fast HPLC Reproducibility
We monitored the reproducibility for a 10 μL injection of loperamide and ketoconazole over a 12.5 hour period in both rat plasma and homogenized rat brain (diluted 4:1 with water). The results are displayed in Figure 5 and demonstrate the overall improved reproducibility that we have observed for ultra fast HPLC. This is important for determining potential matrix ion effects. We have also observed improved carryover in most cases (<0.1 percent) precluding the need for multiple solvent blank injections.
Ultra Fast HPLC Separation
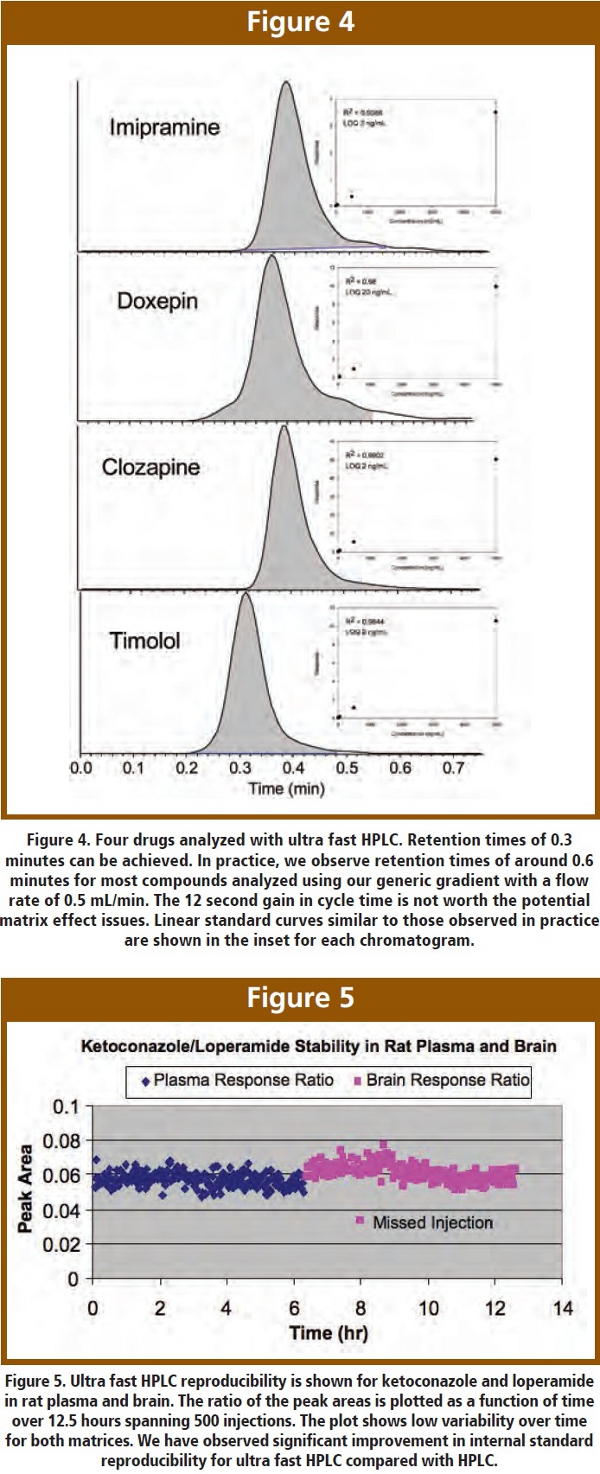
One of the difficulties encountered when screening many compounds is interfering peaks due to endogenous impurities. If the analyte of interest co-elutes with the impurity, quantitation becomes quite challenging. There are various remedies to this problem, such as increasing the LC run to obtain separation, selecting a different fragment ion or improving the sample cleanup (solid phase or liquid-liquid extraction). Unfortunately, these are all “after the fact” solutions and create logistical difficulties in a high throughput arena. Ultra fast HPLC has proven to fix the problem apriori through its improved column efficiency. 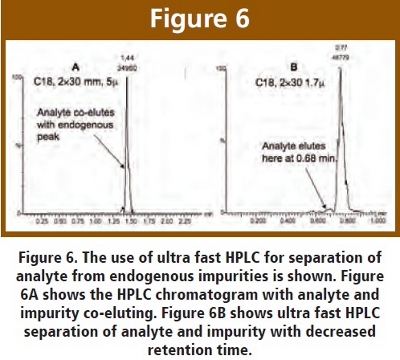 One example is shown in Figure 6, (analyte not shown). An endogenous peak was present at a retention time of 1.44 minutes during a normal HPLC run (2x30mm, 5μm C18). The analyte of interest co-eluted with, and was lost in, this endogenous peak, as shown in Figure 6A. When we switched over to ultra fast HPLC (2x30mm, 1.7μm C18), the endogenous peak eluted at 0.77 minutes. However, the analyte of interest eluted at 0.68 minutes and was sufficiently separated for quantitation. This is clearly a large benefit from ultra fast HPLC, and has saved us on numerous occasions. We routinely operate with ballistic gradients and flow rates of 0.4-0.5 mL/min, taking full advantage of the speed, sensitivity and selectivity of ultra fast HPLC.
One example is shown in Figure 6, (analyte not shown). An endogenous peak was present at a retention time of 1.44 minutes during a normal HPLC run (2x30mm, 5μm C18). The analyte of interest co-eluted with, and was lost in, this endogenous peak, as shown in Figure 6A. When we switched over to ultra fast HPLC (2x30mm, 1.7μm C18), the endogenous peak eluted at 0.77 minutes. However, the analyte of interest eluted at 0.68 minutes and was sufficiently separated for quantitation. This is clearly a large benefit from ultra fast HPLC, and has saved us on numerous occasions. We routinely operate with ballistic gradients and flow rates of 0.4-0.5 mL/min, taking full advantage of the speed, sensitivity and selectivity of ultra fast HPLC.
Ultra Fast HPLC Metabolite Screening
In addition to quantitation of parent drug, there is great utility from obtaining quantitative information about metabolites. This is not straightforward and one must take great care about making quantitative determinations of metabolites when an authentic standard is not readily available. Clearly, within drug discovery it is highly unusual to have anything but the authentic standard of the parent drug. Therefore, in order to obtain quantitative information about metabolites, one must first be sure that a metabolite is being observed. The first criteria is the MS/MS transition. For example, a hydroxyl metabolite will add m/z 16 to the parent molecular weight and may or may not add m/z 16 to the fragment molecular weight (depending on where the modification occurs on the molecule and which piece of the molecule is being observed in the MS/MS transition). If an MS/MS peak is observed that corresponds to a potential metabolite, further confirmation is necessary. Both retention time and MS/MS of the metabolite can serve to authenticate the existence of the suspected metabolite. 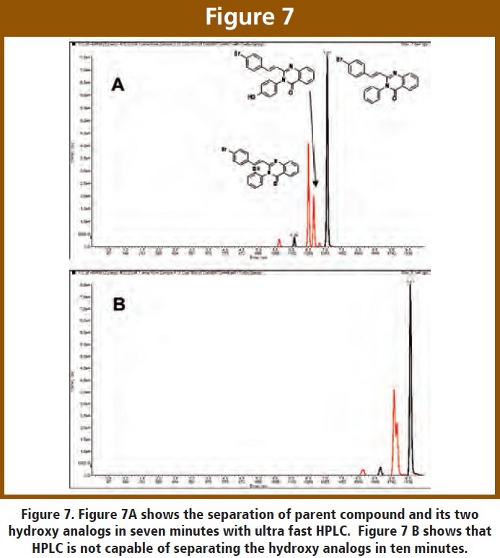 This is where ultra fast HPLC comes into play as shown in Figure 7, for BAS 12439 and two of its hydroxy analogs, CL-3472- 1460 and Chembridge 6141660. Figure 7A shows that ultra fast HPLC can separate the parent as well as both M+16 metabolites in seven minutes while normal HPLC (Figure 7B) would require longer than 9.5 minutes (in practice, this requires at least 20 minutes) to perform the same separation. This is very typical of ultra fast HPLC, and this fast separation represents a breakthrough in metabolite screening [9]. Separation of metabolites from each other, as well as the parent drug, is vital for determining their authenticity. Additionally, we perform MS/MS and MS3 (when needed) on each metabolite in order to further validate their identity as well as characterize their structure. We have arranged our targeted metabolite screening (TMS) paradigm to accommodate every program in conjunction with the CARRS paradigm. Some programs request metabolite information apriori while others may request a specific metabolite following unexpected high or low plasma concentrations of a parent drug.
This is where ultra fast HPLC comes into play as shown in Figure 7, for BAS 12439 and two of its hydroxy analogs, CL-3472- 1460 and Chembridge 6141660. Figure 7A shows that ultra fast HPLC can separate the parent as well as both M+16 metabolites in seven minutes while normal HPLC (Figure 7B) would require longer than 9.5 minutes (in practice, this requires at least 20 minutes) to perform the same separation. This is very typical of ultra fast HPLC, and this fast separation represents a breakthrough in metabolite screening [9]. Separation of metabolites from each other, as well as the parent drug, is vital for determining their authenticity. Additionally, we perform MS/MS and MS3 (when needed) on each metabolite in order to further validate their identity as well as characterize their structure. We have arranged our targeted metabolite screening (TMS) paradigm to accommodate every program in conjunction with the CARRS paradigm. Some programs request metabolite information apriori while others may request a specific metabolite following unexpected high or low plasma concentrations of a parent drug.
The basic problem with quantitation of an analyte without an authentic standard is that the analyte may have a different mass spectrometric response than the surrogate standard. In the case of phase 1 metabolites, such as hydroxylation (M+16) and demethylation (M-14), in which the surrogate standard is the parent drug and the molecular structure is not altered significantly, the difference in response needs to be minimized. In the case of phase 2 metabolites, such as glucuronides, a 10-fold difference in response may be observed, making quantitation in the absence of an authentic standard all but impossible. Acyl glucuronides may be quantified using an indirect approach, as has been described in detail elsewhere [9, 10].
In order to assess the issue of differences in mass spectrometric response between compounds, we investigated a “parent molecule” and a series of hydroxyl group “metabolites” of that molecule. These structures are shown in Figure 2 and include BAS 12439 (parent), CL-3472-1460, Chembridge-6141660, BAS 1578925 and Ibscreen-Stock1S-14627. Figure 8 shows the fragmentation products of the parent molecule BAS 12439. The fragmentation products of the hydroxy “metabolites” are identical besides the addition of m/z 16. Table 3 shows a comparison of the mass spectrometric response of each transition for all five compounds using electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). Several trends are apparent. Firstly, for ESI, mass spectrometric response can vary dramatically. We observed a 2-8-fold response difference between parent and metabolites across all fragments. Ibscreen- Stock1S-14627 had a 10-fold difference in response compared to the other three hydroxy analogs for the m/z 247/263 fragment.  For this molecule, the –OH is on the top (bromine containing) ring of the molecule and is not included in this particular fragment ion whereas it is included in the other two fragment ions (m/z 204/220 and m/z 284/300). It is quickly apparent that, with ESI, it is possible to select a fragment ion that results in dramatic response differences between parent and metabolite. When APCI is used, the situation appears to improve. We observed a 1-4-fold difference in response between parent and analogs across all fragments. Ibscreen-Stock1S-14627 had no more than a 2-fold difference in response compared to the other hydroxy compounds and had the same response as the parent compound for the m/z 247/263 fragment ion. We have tried this approach with a variety of compounds and found that this trend is consistent. While there are certainly exceptions that exhibit a greater than 4-fold response difference, many of the compounds that we examined were within a 2-fold difference for minor molecular modifications under APCI conditions. The bottom line is that, within early discovery, these errors are acceptable for TMS. There has recently been some interest in nano flow LC as a means of normalizing the mass spectrometric response. However, these results are preliminary and require an LC system capable of delivering these low flow rates reproducibly [12].
For this molecule, the –OH is on the top (bromine containing) ring of the molecule and is not included in this particular fragment ion whereas it is included in the other two fragment ions (m/z 204/220 and m/z 284/300). It is quickly apparent that, with ESI, it is possible to select a fragment ion that results in dramatic response differences between parent and metabolite. When APCI is used, the situation appears to improve. We observed a 1-4-fold difference in response between parent and analogs across all fragments. Ibscreen-Stock1S-14627 had no more than a 2-fold difference in response compared to the other hydroxy compounds and had the same response as the parent compound for the m/z 247/263 fragment ion. We have tried this approach with a variety of compounds and found that this trend is consistent. While there are certainly exceptions that exhibit a greater than 4-fold response difference, many of the compounds that we examined were within a 2-fold difference for minor molecular modifications under APCI conditions. The bottom line is that, within early discovery, these errors are acceptable for TMS. There has recently been some interest in nano flow LC as a means of normalizing the mass spectrometric response. However, these results are preliminary and require an LC system capable of delivering these low flow rates reproducibly [12].
We have developed a TMS flowchart that summarizes our “quality control” shown in Figure 9. The basic strategy is straightforward. If the metabolite level is less than 25 ng/mL (as determined by response ratio against analyte standard curve), then it is not of sufficient interest and is not reported. This is determined by using a 25 ng/mL standard of the parent together with a two hour sample. If the metabolite level is greater than 25 ng/mL, it is separated from the parent and other metabolites using a 2.1x50 mm, 1.7-2μm C18 column and MS/MS is performed on the suspected metabolite for verification. We then examine the molecular structure for the possibility of a glucuronide conjugate. Finally, we gather sufficient information to provide the approximate location of the molecular modification.


Conclusion
The advent of 1.7-2μm reproducible HPLC stationary phases and high pressure pumps has opened up a new world of ultra fast HPLC, with a variety of applications ranging from high speed chromatography resulting in greater sensitivity and selectivity to separation of previously unresolved HPLC peaks. We have incorporated this new technology into our daily work and improved turnaround time while increasing the number samples and improving quality. We are currently analyzing 72 compounds per week in the CARRS program, ten compounds per week in the TMS paradigm and nine compounds per week in our new rapid monkey and dog screen. This amounts to around 750 samples spanning close to 100 methods per week. Within one week of sample receipt, 90 percent of these samples are reported.
References
1. Korfmacher WA: Bioanalytical Assays in a Drug Discovery Environment: in Using Mass Spectrometry for Drug Metabolism. W. Korfmacher ed., CRC Press, 2005: 1-34.
2. Cox KA, White RE, Korfmacher WA: Rapid determination of pharmacokinetic properties of new chemical entities: in vivo approaches. Comb. Chem. High Throughput Screen. (2002) Feb;5(1):29-37.
3. White RE: High-Throughput Screening in Drug Metabolism and Pharmacokinetic Support of Drug Discovery. Ann. Rev. Pharmacol. Toxicol. (2000) 40:133–157.
4. White RE, Manitpisitkul P: Pharmacokinetic theory of cassette dosing in drug discovery screening. Drug Metab. Dispos. (2001) 29:957–966.
5. Christ DD: Cassette dosing pharmacokinetics:Valuable tool or flawed science? Drug Metab Dispos. ( 2001) 29:935.
6. Korfmacher WA, Cox KA, Ng K, Veals J, Hsieh Y, Wainhaus S, Broske L, Prelusky D, Nomeir A and White RE: Cassette-accelerated rapid rat screen: A systematic procedure for the dosing and LC-API-MS/MS analysis of new chemical entities as part of new drug discovery. Rapid Commun. Mass Spectrom. (2001) 15:335-340.
7. Mei H, Korfmacher W, Morrison R: Rapid in vivo oral screening in rats: reliability, acceptance criteria, and filtering efficiency. AAPS J. (2006) Jul 28;8(3):E493-500.
8. Dunn-Meynell KW, Wainhaus S, and Korfmacher WA: The use of a ballistic gradient and autosampler method modifications for increasing in-vivo throughput in early drug discovery. Rapid Comm. Mass Spectrom. (2005) 19(20):2905-10.
9. Wang G, Hsieh Y, Cui X, Cheng KC, Korfmacher WA: Ultra-performance liquid chromatography/tandem mass spectrometric determination of testosterone and its metabolites in in vitro samples. Rapid Commun. Mass Spectrom. (2006) 20(14):2215-21.
10. Wainhaus, SB White RE, Dunn-Meynell KW, Grotz D, Weston DJ, Veals J and Korfmacher WA: Semi-quantitation of acyl glucuronides in early drug discovery by LC-MS/MS:, Amer. Pharm. Rev. (2002) 5:86-93.
11. Wainhaus SB: Acyl Glucuronides: Assays and Issues, in Using Mass Spectrometry for Drug Metabolism. W. Korfmacher ed., CRC Press, 2005: 175-202.
12. Utley L, Valaskovic G, Lee MS, Wu J-T: The evaluation of nanospray as a potential response calibrator for the estimation of metabolites levels in drug discovery. 53rd ASMS Conference, Nashville, TN June, 2005.
Dr. Sam Wainhaus is a Group Leader II in the Department of Exploratory Drug Metabolism at Schering–Plough Research Institute. Dr. Wainhaus heads a group of six scientists working on pharmacokinetic and metabolic support of drug discovery. Dr. Wainhaus received his Ph.D. at the University of Illinois at Chicago with Professor Luke Hanley in 1997. He then served as a postdoctoral fellow at Tel Aviv University with Professor Aviv Amirav and at the University of Illinois School of Pharmacy with Professor Richard van Breemen. Dr. Wainhaus joined Walter Korfmacher’s group at Schering-Plough in 1999. Dr. Walter Korfmacher is a Distinguished Fellow, Cymbelene Nardo, Shiyong Wang and Kimberly Dunn-Meynell are Scientist II researchers and Ryan Anstatt is an Associate Scientist within Exploratory Drug Metabolism at Schering- Plough Research Institute.