Introduction
The International Conference on Harmonization (ICH) finalized the ICH Q3D Guideline for Elemental Impurities1 in December 2014. Regulators are now implementing the requirements worldwide. The U.S. Federal Drug Administration2 and the European Medicines Agency3 both adopted a start date of June 2016 for new drug products and December 2017 for authorized drug products. Health Canada has announced that applications submitted after December 31, 2016 must include a risk assessment for elemental impurities. Japanese regulators will begin implementing the guideline for new drug products in April 2017.
The ICH Q3D introduces risk assessment approaches and limits for maximum permitted daily exposure (PDE) based on safety assessments for chronic exposure. However, ICH Q3D does not provide specific daily limits for major components of final drug products, bringing excipients in particular under scrutiny.4 Unlike APIs, excipients do not have established daily doses, leaving manufacturers with less information for calculating concentration limits. This article discusses an ICH Q3D-compliant control strategy model for risk assessment.
Classes of Elements
ICH Q3D classifies 24 elements based on toxicity and likelihood of occurrence in final drug products. Elements in Class 1 are considered a significant risk and may end up in final drug products due to their use in common materials. Elements in Class 2A are considered toxic and are relatively likely to appear in final drug products due to their presence in stainless steel equipment. Class 2B elements are less likely to be incorporated into final drug products, but must be assessed when intentionally added. Class 3 elements are considered non-toxic and must only be evaluated when administered by certain routes. Figure 1 summarizes the elements included in each class, noting when risk assessment is required.
 Figure 1. Elements to be considered in risk assessments.1
Figure 1. Elements to be considered in risk assessments.1ICH Q3D recognizes that other elements – without established PDE levels – may be present in final drug products. These elements (aluminum, manganese, iron, and zinc) may be addressed in other regulations and should be assessed accordingly.
Risk Assessments
Complete risk assessments should consider all sources of elemental impurities including water, manufacturing equipment, process materials, and packaging materials (Figure 2). Some elemental impurities may occur naturally in starting materials from sources that are of mineral or biological origin (i.e., mineral salts from mined sources). Others may be introduced through interaction with manufacturing or container closure systems (i.e., impurities from stainless steel corrosion). Risk assessments should also evaluate packaging materials for liquid drug products and process materials like active charcoal or filter materials.
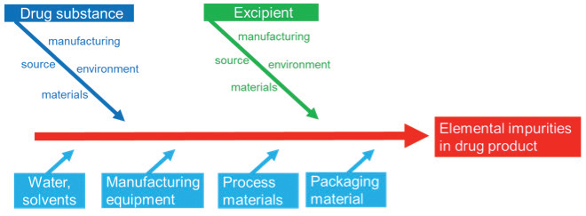 Figure 2. Drug manufacturer’s perspective of possible sources of elemental impurities.
Figure 2. Drug manufacturer’s perspective of possible sources of elemental impurities.Drug product assessment and component assessment approach
ICH Q3D recommends that manufacturers conduct assessments and determine impurity levels using one of two established approaches. In a drug product assessment approach, manufacturers test drug products for the presence of any elemental impurities to support a risk-based control strategy. However, analytical data – without a risk assessment – is not sufficient. Any justification to omit routine controls must have more extensive information than data from limited testing.5
Preferred by manufacturers, the component assessment approach assesses individual components for their contributions to impurities. The combined contribution of an element is compared with the PDE and a control strategy is established, if necessary. When using this approach, drug product manufacturers should ensure that impurities from the manufacturing process are not significant contributors to the overall level of elemental impurities. This approach benefits greatly from supplier information to help analyze data. Figure 3 compares the approaches.
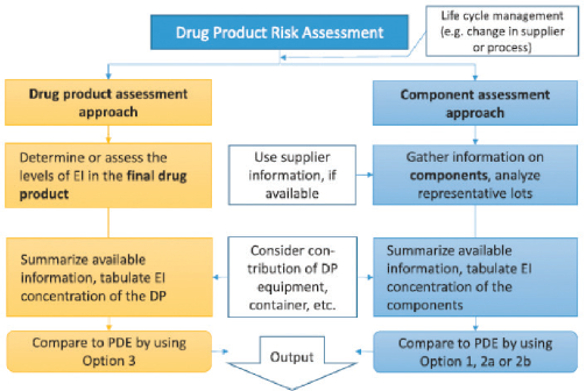 Figure 3. Product and component assessment approach.6
Figure 3. Product and component assessment approach.6Regardless of the approach that manufacturers use, risk assessment will be controlled by life cycle management. Any change in manufacturing process or supplier must be considered and available information should be summarized for easy access and use.
Control Strategy
ICH Q3D provides PDE limits in µg/day for elemental impurities. However, concentration limits in µg/g are more useful for evaluating impurity content in a sample. Chapter 7 of ICH Q3D offers several options for translating between the two:
- Option 1 assumes daily intake of the drug product is 10g (or less)
- Option 2A uses an actual maximum daily intake (versus assuming 10g)
- Option 2B calculates a sum based on known component impurity levels
- Option 3 measures the concentration of elements in the final drug product
Drug products
Not all 24 elements are expected to be detailed in every risk assessment. A number of these elements will be determined as unlikely to be present and, after documentation, no further action is required. Other elements, such as those in Class 1 and Class 2A, need to be considered in all risk assessments. The control threshold is a decision tool that can aid in determining elements at risk of exceeding the PDE. If the observed level of elemental impurity is consistently below the control threshold, defined as 30% of the PDE, existing controls are considered to be adequate.
If the threshold is exceeded, manufacturers should consider additional controls including upstream controls. Alternatively, purification methods may be added at the source of impurities. Specifications should also be established for these elements. For elements exceeding the PDE, a safety assessment can be prepared with a rationale to support higher levels of exposure (i.e., short-term usage, intermittent dosage, a life-threatening disease, etc.). If this additional justification is not available or acceptable, controls should be implemented at the source of contamination, and upstream control or purification methods should be added.
APIs and excipients
While the limits provided by ICH Q3D are specified for final drug products, the same guidelines do not apply to excipients and drug substances. Because excipients do not have a daily dose, there is no established concentration limit, which is generally applicable to excipient manufacturers. Furthermore, compliance of APIs and excipients to a pharmacopoeial substance monograph does not guarantee suitable control of elemental impurities.
As a result, establishing limits for components is a matter of negotiation between drug product manufacturers and their suppliers. To support negotiations, Option 1 (10 g daily dosage) is typically used as the default concentration limit. Please note that the PDEs in ICH Q3D differ depending on the route of administration (i.e., oral, parenteral, inhalation).
If all components have levels below the Option 1 limit for all target elements, these components may be used in any proportion in the final drug product. Figure 4 illustrates this approach.
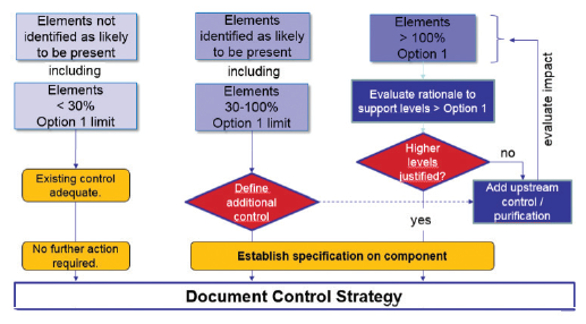 Figure 4. Control strategy for components, based on the Option 1 limit as the default.7
Figure 4. Control strategy for components, based on the Option 1 limit as the default.7Elements intentionally added
Metals or elements intentionally added to the manufacturing process should be considered in every risk assessment. This requires information about which elements have been added, including the relevant production steps and the purge potential of subsequent steps. These elements should be included in the drug substance specification unless the element is controlled by a suitable limit in a synthesis intermediate.
Elements not intentionally added
Elements not added intentionally must also be evaluated. Multi-element analyses are conducted to determine the typical elemental impurity level in APIs, excipients, or chemical process materials.
Figure 5 illustrates an approach to evaluating the results of a multi-element analysis. The y-axis depicts the concentration of the individual elements, the guideline limit, and the respective control threshold. The guideline limit (by default the Option 1 limit) is dependent on the route of synthesis and the maximum daily dose of the drug product. To apply the control threshold as a decision tool, the sources of variability should be understood, both in terms of the analytical method and the variability of the metal level in the specific sources.
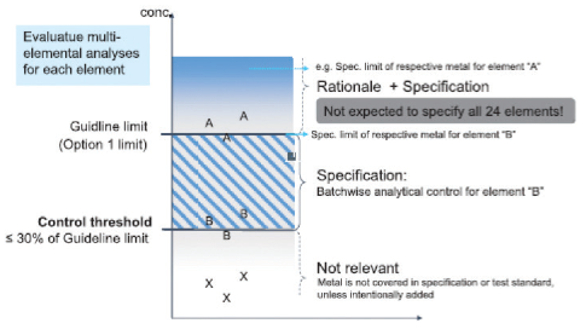 Figure 5. Control strategy for elements not added intentionally.7
Figure 5. Control strategy for elements not added intentionally.7If the results of metal X testing consistently do not exceed the control threshold, the metal can then be excluded from the specification. If metal B is higher than the control threshold, but below the guideline limit, the element should be subject to analytical control and covered in the specification. Any pharmaceutical substance with element results higher than the guideline limit (Option 1 limit by default) for the drug product may still be used in final drug products. However, a rationale will be appropriate to support higher levels. For pharmaceutical substances at this level, a suitable specification limit should be defined.

Conclusion
The implementation of ICH Q3D is an important milestone to harmonize control of elements worldwide. While these guidelines set strict limits for final drug products, limits for excipients, APIs, and other pharmaceutical substances are not included. With the component assessment approach, the contribution of elemental impurities of each component is assessed. The option 1 limit (based on 10 g daily dose) may be used as default concentration limit. This approach allows manufacturers to provide crucial information about the contribution of impurities in final drug products from excipients or APIs.
References
- ICH. 2014. ICH Harmonized Guideline Q3D, Guideline for Elemental Impurities, Step 4.
- US FDA. June 2016. Elemental Impurities in Drug Products. Draft Guidance.
- EMA. 2015. Elemental impurities in marketed products: Recommendations for implementation. EMA/CHMP/QWP/109127/2015.
- Teasdale, A et. al. 2015. Implementation of ICH Q3D Elemental Impurities Guideline: Challenges and Opportunities. Pharm. Technol. 39(3):36-49.
- EMA. 2016. Implementation strategy of ICH Q3D Guideline. Draft. EMA/404489/2016.
- ICH. 2015. Q3D training module 5: Risk Assessment
- Reichert, U. 2016. Elemental Impurities: ICH Q3D finalized. Pharm. Ind. 78(5): 670-676