Introduction
Biopharmaceutical products are now main players within the pharmaceutical industry. However, many biological and biotech products are inherently labile compared to small molecule APIs and so liquid formulations may not always deliver suitable stability for long shelf life and ease of global distribution. Therefore, the dried state is an attractive option, although the drying process is not without its own challenges. In particular, the stripping of the native water shell from proteins may result in loss of potency and denaturation and so the appropriate choice of lyoprotectant excipients is required.
Lyophilization is a key process step in the manufacture of many biopharmaceuticals and as these products are of increasing importance in the product portfolio of pharmaceutical manufacturers, it continues to be used as a vital unit step in bioprocessing. However, it is a very expensive processing step, requiring large capital outlay for the equipment and ancillary services, and has lagged behind other processes stages such as chromatography and large scale filtration in terms of the interest shown by chemical engineers to model and optimize it. Historically, freeze drying cycles were derived by trial and error and once established and validated, pharmaceutical freeze drying processes would be inflexible and often time-consuming, while the underlying physical processes remained little understood. This often resulted in inefficient and unwieldy processes, with process changes to licensed products being expensive and time-consuming to introduce. Although some groups were seeking to provide a greater technical understanding of the lyophilization process, much of this was based at laboratory and pilot scale, and the major innovations in equipment manufacture over the past 20 years were focused mainly on larger and more automated freeze-drying plant and associated delivery systems such as autoloaders, isolators, etc. The issue of process scale up would be a major challenge as failure at large scale would be expensive and delay new product development.
With the paradigm changing introduction of the Quality-by-Design model [1], commenced by the FDA in the first decade of the 21st century, greater flexibility became possible as the emphasis was on a greater understanding of the manufacturing processes and the biophysical properties of the product. This would result in the construction of a design space which facilitates scale up, site transfer and process change. The establishment of the Design Space, as defined in the ICH Q8 guidance [2], requires appropriate use of sensitive discriminating validated analytical techniques and a greater understanding of the key engineering processes which underly the manufacturing process [3]. Although beginning with other key bioprocesses, lyophilization has now become a target for study, and the application of QbD to lyophilization has become a popular topic [4], that with the development of appropriate technology is an achievable goal [5] .
Review of PAT Tools & Predictive/Monitoring Processes Used in Lyophilization
Lyophilization comprises three main elements: freezing - which allows for complete solidification of the material to be dried; primary drying or sublimation - which accounts for the removal of 90% of the water content; and secondary drying or desiccation - which accounts for the remainder of the water to leave a product with a target moisture of 0.5-3%w/w. Process monitoring of lyophilization with product thermocouple probes has been used for many years but only a few vials can be monitored this way and it is well documented that they frequently behave anomalously with regards to the rest of the batch. The development of wireless temperature sensors [6,7] has addressed this to some extent but there is still the need to monitor the whole batch and not just a few individual vials. Gravimetric measurement of water loss during sublimation has been used to monitor the process, microbalances have been described to monitor individual containers, and one-off builds have allowed for measurement of water loss on multiple containers in pilot units, although practically this has been only applicable in small scale dryers or off-line to assess sublimation rates through truncated trial sublimation runs. Pressure rise measurements, where the isolator valve is closed and the rise in vapor pressure resultant from sublimation process is measured, were introduced in the 1990s [8-10] and are a means of measuring the sublimation rate for the whole batch in the drying chamber. It has gained acceptance over a number of years, though may be less applicable where the isolator valve is slow to respond (particularly true of large freeze dryers) and of course can not be used where the condenser is integral. More recently, vapor flow monitors such as the tunable diode laser absorbance spectroscopy (TDLAS), monitoring water vapor flux from chamber to condenser, has been heralded as a major step towards effective monitoring of the drying process [11,12] and other techniques such as plasma emission spectrometry measuring moisture content within the chamber have also been reported [13]. Most recently near-infrared and Raman spectroscopy with fibre optic probes have introduced additional product characterization although applicable only to laboratory/pilot scale freeze dryers [6]. Several recent papers have advocated the advantages of combining several techniques and the work of De Beer et al [14] has shown the power of this approach by following both nucleation, water sublimation and crystallization changes in excipient as well as by monitoring primary and secondary drying endpoints.
Alongside the development of such analytical methods has come the development of mathematical models of the lyophilization process. Although drying and sublimation rates were discussed at length in older works (e.g. Mellor [15]), it was the paper of Pikal et al [16] which brought pharmaceutical attention to the impact of heat and mass transfer coefficients in sublimation. Pseudo steady-state moving planar boundary models for sublimation have been developed by Pikal and others, such as two-dimensional axis-symmetrical unsteady state models, have also been derived [17]. The sublimation in edge vials where radiant heat may have an influence (especially in pilot dryers), may be considered, in addition to those in the center of shelves that are shielded from such influences [18]. However, to be practical in control as well as monitoring applications of the drying process, more simple models must be applied [19]. The use of manometric temperature modelling (MTM) tests in their research papers has found practical application in the SMART technology which have been used to predict cycles based upon the known properties of the formulation (e.g resistance of the dried layer), and dimensions and thermal properties of the containers [20]. Primary drying end point could also be determined by the comparison of the chamber pressure as measured by Pirani and capacitance gauges whose sensitivity differ when water vapor is subliming and when it has effectively completed. Monitoring based on a combination of soft-cell sensors (smart vials) and pressure rise testing have been presented by Barresi et al [7,21], here the predicted sublimation model can be updated in real time with successive pressure rise results so that a single run may be sufficient to derive a satisfactory optimized cycle. The use of TDLAS again allows interactive monitoring and modification of the cycle run in response to the progression of the sublimation process. The increasing impact of product dry layer resistance during processing needs to be taken into account [22].
Although these technologies have been applied in some cases to pilot units, only some may be applicable in true production scale units, also allowing the possibility of on-the-fly process control at production scale. The types of cycle derived from these methods are very different to the traditional ones with shelf temperature at the start of cycle often being relatively high and falling as primary drying completes [23].
Most effort has been applied to the optimization of primary drying as this is usually the longest step and the one during which product collapse is most likely to occur. However, some work on optimization of secondary drying has been done, especially with TDLAS [24]. Most recently, focus has again been drawn to the freezing stage and the inherent variability in drying due to stochastic ice nucleation and the resultant size of ice crystals. The impact of ice crystal size and the benefits of annealing steps to address the ice crystal size was reported by Searles et al in 2001 [25] but the inherent stochastic nature of the nucleation event in any given vial has been a significant problem. However, several recent papers have reported the controlled nucleation of ice in vials in a lyophilizer and the advantages this would bring to shortening process cycles [26,27].
Rationale of the Microscale Down Approach to Downstream Processing
The miniaturization of large-scale process operations enables process understanding to be gained more rapidly and cost effectively than by sequential pilot-scale runs, due to increased parallelization and through the use of smaller quantities of materials. Precise miniaturization of a process step is not always possible because physical and thermodynamic effects such as mixing and heat transfer do not simply scale down in proportion to vessel geometry. However, ultra scale-down (USD) studies can be used to mimic process parameters that are critical to performance in large scale processes, while using significantly smaller quantities of material. For example, bespoke USD devices have been designed and developed to mimic bioreactors for fermentation [28-30], shear effects during centrifugation [31] and mixing effects in protein dilution refolding [32], while miniature columns [33] or resin loaded pipette tips [34] can be used to predict the performance of large- scale chromatography.

Figure 1. Sublimation rate per unit area versus ratio of contact area: volume, for vials, ampoules and microwells (contact area being that in thermal contact with the shelf).
In addition to USD using bespoke device designs, simple microtitre plates can also facilitate multiple parallel experimentation that is readily automated with liquid handling robotics that improve accuracy. This can be used for scouting bioprocess conditions, as well as for detailed bioprocess characterization and modelling as demonstrated for fermentation [35,36], soluble protein expression in E. coli [37], cell lysis [38], microfiltration [39], protein refolding from inclusion bodies [40,41], protein precipitation [42-44], chromatographic resin screening [45], protein crystallization [46], and freeze-drying as discussed in more detail below, as well as for measuring the effects of additives, ligands or mutations upon the stability of proteins [47-50].
Miniaturization has continued further to enable bioprocess evaluation and analysis of biological materials in microfluidic devices that use only nanolitre to low microlitre volumes of material. These include miniature bioreactors for fermentation [51], microfluidic chromatography columns [52,53] , enzyme reactors [54,55], and for the measurement of protein stability and ligand interactions [56].
Recently, in a joint project between The Advanced Centre for Biochemical Engineering, University College London and NIBSC, we have investigated the benefi ts of applying a Design of Experiment (DoE) approach to formulation optimization, in combination with freeze-drying within microplates. Using a microtitre plate format, the amounts of active biological material required can be minimized. This might have particular benefi ts where the available quantity of a biological material is at a premium, such as in preclinical development, while still enabling a broad sweep analysis of potential excipients to be conducted. Others have considered the use of V-well plates [57], and a commercial microtube format has also become more recently available. These require a specialized metal support plate to ensure good thermal contact with the shelf. In our approach the microplates were modified to ensure direct contact with the base of the flat-well plates and the shelf of the freeze dryer, although a fl at metal plate could also potentially be inserted to thermally bridge the gap to the shelf. The diameter of each microplate well is constant, which closely mimics the geometry of glass vials, unlike the V-well plate. The thermal properties of the plastic plates were also compared to those of the glass vials and ampoules currently in use at NIBSC and shown to be linearly related. Although sublimation rates were somewhat higher in the plastic wells, they were considered sufficiently similar to allow a valid comparison of formulants to be performed (Figure 1) . Consideration of edge effects, commonly seen in microtitre applications was also made and the edge wells were not used in subsequent studies, but were filled with water or buffer.
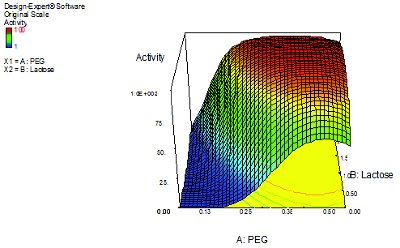
Figure 2. Microtitre DoE study: contour map of optimization of retained LDH activity (% of the pre-freeze dried activity) in terms of PEG and lactose concentrations (% w/w).
In a published study [58] of a model protein – lactic dehydrogenase, LDH, a tetrameric protein known to be unstable to freeze drying, we have demonstrated that a rapid two-stage DoE approach was suffi cient to screen a series of formulants for their impact on preserving LDH enzymic activity. A fractional factorial design was performed to assess the impact of 7 common freeze-drying excipients, from which lactose and polyethylene glycol (PEG) were chosen for further study based upon the evidence of a half-normal probability plot of the impact of each excipient on the retention of activity. A central composite face DoE phase of optimization was then undertaken and a contour plot developed that mapped the effect of concentration for the selected excipients upon retention of activity after freeze-drying (Figure 2). The optimal formulation was then trialed in a conventional vial format and the improved retention of biological activity confi rmed under standard freeze- drying conditions.
This approach required fewer experiments than a one-factor-at-a- time (OFAT) study and was achieved using only tens of micrograms of enzyme. This approach has since been applied to a clinically relevant biotechnology product with similarly encouraging outcome [59]. Although DoE is widely used and accepted in drug formulation development [60], the combination of this with freeze drying in the microtitre format allowed for greater throughput as multiple 96-well plates could be freeze dried on a single shelf and so a wide number of options could be screened simultaneously Design of Experiment Used in Lyophilization Cycle Development

Figure 3. Influence of factors on cake acceptability ordered by modulus of co-efficient. The factors that have the highest impact on cake acceptability are freezing temperature, then primary drying pressure & temperature and secondary drying temperature (where T, t and P are Temperature, time and Pressure & F ,PD, SD are freezing, primary drying & secondary drying phases)
Having optimized the formulation for LDH in the earlier study, we considered applying Design of Experiment technique to optimize the freeze drying cycle for a 1ml fill in 22 mm diameter type 1 glass vials. Rather than performing a model of sublimation rate or study individual stages of the process it was decided to model the entire process. Residual moisture content (quantitative categorical) and cake appearance (qualitative categorical,0 =visually unacceptable , 1= visually acceptable) were identified as critical outputs and modulated differential scanning calorimetry used to identify the glass transition temperature of the formulation (0.05U/ml LDH in 1.7%w/v lactose, 0.41% PEG in 50mM sodium phosphate buff er pH 7) as -16°C. The parameters chosen were freezing temperature (ranging from -40 to -21°C) and freeze length (30 to 240 min), primary drying shelf temperature (from -21 to -11°C) and duration (from 360-720 min), chamber vapor pressure (from 10 to 60% of the saturated vapor pressure of ice at the set shelf temperature). Secondary drying parameters were set as temperature 0 to 30°C, duration 0-720min and pressure from 30-100mTorr. Ramps were set as maximum rate for the dryer and 10 vials were dried in each experiment surrounded by empty vials so as to minimize any shelf edge or radiative heat eff ect. A central composite face design was applied and a total of 60 runs were performed. Freeze dried vials from runs were tested for appearance, residual moisture and also functional activity. A highly complex multifactorial equation was derived for each categorical that related the final residual moisture to the operational parameters. When the impact of each factor was assessed and ranked, freezing temperature had by far the greatest impact on cake acceptability with primary drying pressure and temperature and secondary drying temperature being ranked next (Figure 3). Only 5 of the factors had a significant impact on residual moisture categorical, the greatest unsurprisingly being the length of secondary drying (but not secondary drying chamber pressure in line with published findings of Nail et al [61]) and the chamber pressure of the primary drying.
Equation of Model Cake acceptability = 4.56 + 0.2TF – (2.76x10-3)tF + 0.01TPD + (5.02x10-3)tPD -0.05PD – 0.02TSD- (2.28x10-4)tSD- (2.52x10-3)PSD + (4.56x10-5)TFPPD + (4.9x10-5)TFPSD - (5.83x10-5)TPDtSD + (3.42x10-4)PPDTSD - (8.35x10-6)tSDPSD + (3.04x10-3)TF2 + (4.14x10-4)PPD2
Where T, t and P are Temperature, time and Pressure & subscripts F, PD, SD are freezing primary drying & secondary drying phases.
The usefulness of the DoE experiment was that the key parameters affecting the critical quality attributes were identified. DoE could be used further by keeping certain sections common (for instance the secondary drying or freezing conditions) to allow the impact on the various operational parameters of an individual stage e.g. primary drying to be identified.
Future Prospects
In order to speed up formulation development for freeze drying and begin initial studies of likely rate limiting steps DoE and the microscale down studies described may be able to help. We have recently been applying DoE studies in a range of containers (vials and ampoules) and formulations in order to better understand the impact of formulation and container surface area on sublimation rates. Bhamabani & Medi [62] reviewed the importance of the effects of container /closures in lyophilization applications. We have reviewed too how new PAT technologies are delivering powerful new methods to assist in lyophilization cycle optimization and understanding of the physical state of the product during the process [63]. However, delivery of an effective laboratory or pilot scale lyophilization cycle is only part of the task.
One of the greatest problems in freeze drying is scale up from laboratory cycles, which may work well, to huge production scale freeze dryers where the same conditions may be unsuccessful. The factors bedevilling scale up have been well documented. For aggressively optimized cycles choked flow may be an issue and indeed vapor flow dynamics have shown that inter-shelf spacing may also be a limitation [64]. The impact of partial against complete dryer loads were investigated by Patel et al [65]. However the use of an array of PAT techniques and DoE strategies to better understand the Design Space can produce a more robust cycle to meet process scale demands [66].
Acknowledgements
The authors thank the Engineering & Physical Sciences Research Council (EPSRC, UK) and NIBSC for funding an engineering doctoral studentship to YG.
References
- FDA(2007) Pharmaceutical Quality for the 21st Century: A Risk-Based Approach http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm128080.htm
- International Congress on Harmonization (2008) Pharmaceutical development Q8(r1)ICH tripartite Guideline. http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html
- Jameel F & Kahn M (2009) Quality by Design as applied to the developing & manufacturing of a lyophilised protein product. Amer Pharm Rev 12; Nov 2009 pp20-4.
- Nail S & Searles J(2008) Elements of Quality by Design in development & scale up of freeze dried parenterals. Biopharm Int 21; 44-52.
- Guttzeit M (2010) Designing an effective PAT driven scale up of lyophilization processes. Pharm Tech Europe 22;30-6.
- De Beer TRM, Wiggenhorn M, Veilon R, Debacq C, Mayeresse Y, Moreau B, Burggraeve A, Quinten T, Friess W, Winter G, Vervaet C, Remon JP, Baeyens WRG.(2009)Importance of using complementary process analysers for the process monitoring analysis & understanding of freeze drying. Anal Chem 81; 7639-49.
- Barresi AA, Velardi SA, Pisano R, Rasetto V, Vallan A Galan M (2009) In line control of the lyophilization process - a gentle PAT approach using software sensors Int J Refrig 32; 1003-14.
- Milton N, Pikal MJ, Roy ML et al (1997) Evaluation of a manometric temperature measurement as a means of monitoring product temperature during lyophilization PDA J Pharm Sci Technol 5;7-16.
- Oetjen GW (1999) “Freeze Drying” Weinheim, Wiley VCH, 1999.
- Liapis AI, Sadikoglu H (1998) Dynamic pressure rise in the drying chamber as a remote sensing method for monitoring the temperature of the product during the primary drying stage of freeze drying. Drying Technol 15;1153-71.
- Gieseler H, Kessler W , Finson M et al (2007) Evaluation of tunable diode laser absorption spectroscopy for in process water vapor mass flux measurements during freeze drying . J Pharm Sci 96;1776-93.
- Kuu WY & Nail SL (2009) Rapid freeze drying cycle optimization using computer programs developed based on heat and mass transfer models and facilitated by TDLAS. J Pharm Sci 98:3469-82.
- Mayeresse Y , Veillon R, Sibille PH, Nomine C (2007) Freeze drying process monitoring using a cold plasma ionisation device. PDA J Pharm Sci Technol 61; 160-74.
- De Beer TRM, M Wiggenhorn, Hawe A, Kasper JC, Almeida A , Quinten T, Friess W, Winter G, Vervaet C, Remon JP. (2011) Optimization of a pharmaceutical freeze dried product & its process using an experimental design approach & innovative process analysers. Talanta 83;1623-33.
- Mellor JD (1978) “Fundamentals of Freeze Drying “, Academic Press, London.
- Pikal MJ, Roy ML, Shah S (1984) Heat and mass transfer in vial freeze drying of pharmaceuticals, the role of the vial. J Pharm Sci 73; 1224-37.
- Kramer T,Kremer DM, Pikal MJ, Petre WJ, Shalaev RY, Gatlin LA (2009) Procedure to optimize scale up for the primary drying phase of lyophilization . J Pharm Sci 98;307-18.
- 1Brulls M & Rasmusson A (2009) Ice sublimation in vial lyophilization. Drying Technol 27;695-706.
- Pisano R, Fissore D, Barresi AA (2011) Freeze drying cycle optimization using predictive control techniques. Indust Eng Chem Res 50;7363-79.
- Tang XC, Bail SL, Pikal MJ (2005) Freeze drying process design by manometric temperature measurement: design of a smart freeze dryer . Pharm Res 22;685-700.
- Fissore D, Pisano R,Velardi S,Barresi A Galan M (2009) PAT Tools for the optimization of the freeze drying process Pharm Eng 29(5); pp 58-68
- Mockus L, LeBlond D, Basu PK, Shah RB, Khan MA (2011) QbD case study Bayesian prediction of lyophilization cycle parameters. AAPSPharmSciTech 12; 442-8.
- Pisano R, Fissore D, Velardi SA, Barresi AA. (2010) Inline optimization & control of an industrial freeze drying process for pharmaceuticals J Pharm Sci 99; 4691-4709.
- Schneid SC, Gieseler H, Kessler WJ, Luthra SA, Pikal MJ (2011) Optimization of the secondary drying sterp in freeze drying using TDLAS technology . AAPS PharmSciTech 12;379-87.
- Searles J, Carpenter J, Randolph T (2001) Ice nucleation temperature determine the primary drying rate of lyophilization for samples frozen on a temperature controlled shelf. J Pharm Sci 90;860-71.
- Patel SM, Bhugra C, Pikal MJ (2009) Reduced pressure ice fog technique for controlled ice nucleation during freeze drying. AAPS PharmSciTech10;1406-11.
- AK Konstantinidis AK, Kuu W, Otten L, Nail SL, Sever RR (2011) Controlled nucleation in freeze drying: effects on pore size in the dried product layer, mass transfer resistance & primary drying rate. J PharmSci 100;3453-70.
- Betts, J. I., Doig, S. D., Baganz, F., 2006. Characterization and application of a miniature 10 mL stirred-tank bioreactor, showing scale-down equivalence with a conventional 7 L reactor. Biotechnol. Prog 22, 681-688.
- Reis, N., Goncalves, C., Vicente, A., Teixeira, J., 2006. Proof-of-concept of a novel micro-bioreactor for fast development of industrial bioprocesses. Biotechnol. Bioeng. 95, 744-753.
- Walther, I., Vanderschoot, B. H., Jeanneret, S., Arquint, P., Derooij, N. F., Gass, V., Bechler, B., Lorenzi, G., Cogoli, A., 1994. Development of A Miniature Bioreactor for Continuous-Culture in A Space Laboratory. J. Biotechnol. 38, 21-32.
- Boychyn, M., Yim, S. S. S., Bulmer, M., More, J., Bracewell, D. G., Hoare, M., 2004. Performance prediction of industrial centrifuges using scale-down models. Bioprocess and Biosystems Engineering 26, 385-391.
- Mannall, G. J., Titchener-Hooker, N. J., Dalby, P. A., 2007. Factors affecting protein refolding yields in a fed-batch and batch-refolding system. Biotech Bioeng 97, 1523-1534.
- Hutchinson, N., Chatre, S., Baldascini, H., Davies, J., Bracewell, D., Hoare, M., 2009. Ultra Scale-Down Approach to Correct Dispersive and Retentive Effects in Small-Scale Columns When Predicting Larger Scale Elution Profiles. Biotechnol. Prog 25, 1103-1110
- Wenger, M. D., Dephillips, P., Price, C. E., Bracewell, D. G., 2007. An automated microscale chromatographic purification of virus-like particles as a strategy for process development. Biotechnology and applied Biochemistry 47, 131-139.
- Doig, S. D., Pickering, S. C. R., Lye, G. J., Baganz, F., 2005. Modelling surface aeration rates in shaken microtitre plates using dimensionless groups. Chem Eng Sci 60, 2741-2750.
- Duetz, W. A., 2007. Microtiter plates as mini-bioreactors: miniaturization of fermentation methods. Trends Microbiol. 15, 469-475.
- Islam, R. S., Tisi, D., Levy, M. S., Lye, G. J., 2007. Framework for the rapid optimization of soluble protein expression in Escherichia coli combining microscale experiments and statistical experimental design. Biotechnol. Prog. 23, 785-793.
- Wenger, M. D., DePhillips, P., Bracewell, D. G., 2008. A microscale yeast cell disruption technique for integrated process development strategies. Biotechnol. Prog. 24, 606-614.
- Jackson, N. B., Liddell, J. M., Lye, G. J., 2006. An automated microscale technique for the quantitative and parallel analysis of microfiltration operations. J Membrane Sci 276, 31-41.
- Mannall, G. J., Myers, J. P., Liddell, J., Titchener-Hooker, N. J., Dalby, P. A., 2009. Ultra scale-down of protein refold screening in microwells: Challenges, solutions and application. Biotechnol. Bioeng. 103, 329-340.
- Merli, S., Corti, A., Cassani, G., 1995. Production of soluble tumor necrosis factor receptor type I in Escherichia coli: optimization of the refolding yields by a microtiter dilution assay. Anal. Biochem. 230, 85-91.
- Ahmad, S. S., Dalby, P. A., 2011. Thermodynamic parameters for salt-induced reversible protein precipitation from automated microscale experiments. Biotechnol. Bioeng. 108, 322-332
- Kramarczyk, J. F., Kelley, B. D., Coffman, J. L., 2008. High-throughput screening of chromatographic separations: II. Hydrophobic interaction. Biotechnol. Bioeng. 100, 707-720.
- Wiendahl, M., Voelker, C., Husemann, I., Krarup, J., Staby, A., Scholl, S., Hubbuch, J., 2009. A novel method to evaluate protein solubility using a high throughput screening approach. Chem Eng Sci 64, 3778-3788.
- Coffman, J. L., Kramarczyk, J. F., Kelley, B. D., 2008. High-throughput screening of chromatographic separations: I. Method development and column modeling. Biotechnol. Bioeng. 100, 605-618.
- Knevelman, C., Davies, J., Allen, L., Titchener-Hooker, N. J., 2010. High-Throughput Screening Techniques for Rapid PEG-Based Precipitation of IgG(4) mAb from Clarified Cell Culture Supernatant. Biotechnol. Prog 26, 697-705.
- Aucamp, J. P., Cosme, A. M., Lye, G. J., Dalby, P. A., 2005. High-throughput measurement of protein stability in microtiter plates. Biotechnol. Bioeng. 89, 599-607.
- Aucamp, J. P., Martinez-Torres, R. J., Hibbert, E. G., Dalby, P. A., 2008. A microplate-based evaluation of complex denaturation pathways: structural stability of Escherichia coli transketolase. Biotechnol. Bioeng. 99, 1303-1310.
- Mahendrarajah, K., Dalby, P. A., Wilkinson, B., Jackson, S. E., Main, E. R., 2011. A high-throughput fluorescence chemical denaturation assay as a general screen for protein-ligand binding. Anal. Biochem. 411, 155-157.
- Matulis, D., Kranz, J. K., Salemme, F. R., Todd, M. J., 2005. Thermodynamic stability of carbonic anhydrase: Measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 44, 5258-5266.
- Szita, N., Boccazzi, P., Zhang, Z. Y., Boyle, P., Sinskey, A. J., Jensen, K. F., 2005. Development of a multiplexed microbioreactor system for high-throughput bioprocessing. Lab on A Chip 5, 819-826
- Darton, N., Reis, N., Mackley, M., Slater, N., 2011. Fast cation-exchange separation of proteins in a plastic microcapillary disc. J Chromatogr A 1218, 1409-1415.
- Shapiro, M. S., Haswell, S. J., Lye, G. J., Bracewell, D. G., 2009. Design and Characterization of a Microfluidic Packed Bed System for Protein Breakthrough and Dynamic Binding Capacity Determination. Biotechnol. Prog 25, 277-285.
- Matosevic, S., Lye, G., Baganz, F., 2011. Immobilised enzyme microreactor for screening of multi-step bioconversions: Characterization of a de novo transketolase-omega-transaminase pathway to synthesise chiral amino alcohols. J. Biotechnol. 155, 320-329.
- Ngamsom, B., Hickey, A. M., Greenway, G. M., Littlechild, J. A., Watts, P., Wiles, C., 2010. Development of a high throughput screening tool for biotransformations utilising a thermophilic L-aminoacylase enzyme. J. Mol. Catal. B: Enzym 63, 81-86.
- Gaudet, M., Remtulla, N., Jackson, S. E., Main, E. R., Bracewell, D. G., Aeppli, G., Dalby, P. A., 2010. Protein denaturation and protein:drugs interactions from intrinsic protein fluorescence measurements at the nanolitre scale. Protein Sci. 19, 1544-1554.
- Von Graberg S & Gieseler H (2006) Freeze drying in non-vial containers - evaluation of heat transfer coefficients of PCR paltes and correlation to freeze drying cycle. Freeze drying of Pharmaceuticals & Biologicals, Garmisch Partenkirchen ,Germany, Oct 3-6, 2006. Available at http:www.freeze-drying.eu/add_doc/pub/_6.pdf
- Grant Y, Matejtschuk P, Dalby PA (2009) Rapid optimization of protein freeze-drying formulations using ultra scale-down and factorial design of experiment in microplates. Biotech Bioeng 104;957-64.
- Grant Y, Matejtschuk P, Bird C, Wadhwa M, Dalby PA (2012) Freeze drying formulation using microscale and design of experiment approaches: a case study using granulocyte colony-stimulating factor. Biotechnol Lett 34; 641-8.
- Capelle MAH, Gurny R, Arvinte T. (2007) High throughput screening of protein formulation stability : practical considerations Eur J Pharm Biopharm 65; 131-48.
- Nail S, Jiang S, Chongprasert S , & Knopp S (2002) Fundamentals of Freeze Drying in “Development and manufacture of protein pharmaceuticals” Steven Nail & Michael J.Akers, eds., Plenum , New York, pp. 281-359.
- Bhamabani A & Medi BM (2010) Selection of container/closures for use in lyophilization applications. Amer Pharm Rev 13;86-91.
- Krishnan S, Cao, W, Phillips J (2009) Applying Raman spectroscopy to design of lyophilization cycles for protein formulation development. Amer Pharm Rev 12; 44-53.
- Sane SV & Hsu CC (2007) Strategies for successful lyophilisation process scale up Amer Pharm Rev 10;132-6
- Barresi AA, Fissore D, Marchiso DL (2010) Process Analytical Technology in Industrial Freeze drying in “Freeze drying/Lyophilization of Pharmaceutical & Biological Products “ ed Rey L & May JC , 3rd ed. pp460-93. Informa Healthcare,New York
- Patel SM,Jameel F ,Pikal MJ (2010) The effect of dryer load on freeze drying process design. J Pharm Sci 99;4363-79.
- Sundaram J, Max Shay YH, Hsu CC, Sane SU (2010) Design space development for lyophilization using Design of Experiment and process modelling. Biopharm Int 23;26-36.
Author Biographies
Yitzchak Grant is currently working as a Senior Process Engineer at Kraft Foods, Banbury, UK. He has a 1st class honours BEng(2006) from University College London where he also successfully studied for an Eng Doc under the supervision of his co-authors. He has pharmaceutical experience work at MedImmune(Cambridge, UK) and Pfizer(Sandwich, UK).
Paul Dalby (MA, Ph.D., University of Cambridge) is a Reader in Biochemical Engineering and Biotechnology at University College London, where he combines protein engineering biophysics and high-throughput screening technology development, to improve therapeutic protein bioprocessing.
Paul Matejtschuk (Ph.D., University of Warwick, UK) is a Principal Scientist, leading the Standardization Science Section, responsible for formulation and freeze drying development at NIBSC, a Centre of the UK’s Health Protection Agency, and has many years postdoctoral experience in the processing and characterization of therapeutic and diagnostic proteins.