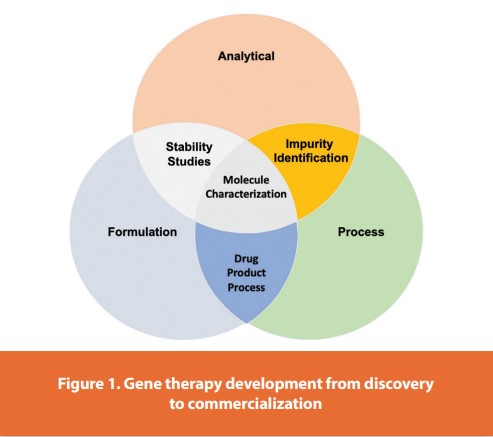
The field of clinical gene therapy has advanced rapidly and now accounts for over 2,200 clinical trials initiated since 1989, of which ~65% are being conducted in the USA.1 Progress in the field has contributed to a new generation of gene therapy commercial initiatives, including several startup companies. Since the beginning of 2013, at least $600 million of venture capital funding has been raised to support gene therapy, a strong comeback after several years of slow progress.2,3 The estimated global gene therapy market value is estimated to exceed $10 billion by 2025.4,5 In October of 2012, the European Medicines Agency approved the use of a gene therapy product (Glybera) for the treatment of adult patients diagnosed with familial lipoprotein lipase deficiency.6 The approval comes nine years after the State Food and Drug Administration of China (SFDA; Beijing, China) approved the world’s first gene therapy product (Gendicine) for the treatment of head and neck squamous cell carcinoma in 2003.7 This was followed in 2005 by the approval of Oncorine by the SFDA, the first commercialized oncolytic adenovirus for patients with late-stage refractory nasopharyngeal cancer8 and the approval of Rexin-G, a tumor-targeted gamma-retroviral vector expressing cytocidal cyclin G1 by the Philippine FDA in 2007.9 Also, the Russian Ministry of Healthcare and Social Development issued a market authorization in 2013 for Neovasculgen, a plasmid vector that expresses Vascular Endothelial Growth Factor gene for the treatment of peripheral arterial disease through angiogenesis.10 In October 2015, positive results from a Phase III clinical trial for LCA2 (meeting its primary endpoints) were reported by Spark Therapeutics.11 These results led to the first commercial product to be approved in the USA,12 “History is made by FDA approves Spark’s Vision-Restoring Gene Therapy”. On October 12, 2017; a FDA Advisory Committee voted 16 to 0 in recommending marketing approval for LUXTURNA.47 The acceleration in the total number of gene therapy clinical trials, as well as the increased number of later-stage clinical trials, signals an exciting era that promises to lead to the emergence of this new therapeutic paradigm for previously unmet therapeutic needs. The increased interest also stimulated the emergence of several new gene therapy companies, many closely affiliated with the academic centers that fostered the technology.13 This trend has led to a greater demand for both pre-clinical and clinical-grade viral vector manufacturing capacity to support the increasing number of gene therapy clinical development programs. As these programs advance towards licensure, more rigorous product characterization using improved analytical methods, and progressively higher regulatory compliance will be required. Herein, highlight several significant clinical successes in the field of gene therapy and provide examples of challenges in vector manufacturing, vector characterization, and compliance. Gene therapy development from discovery to commercialization, Figure 1.
Cell Line Development
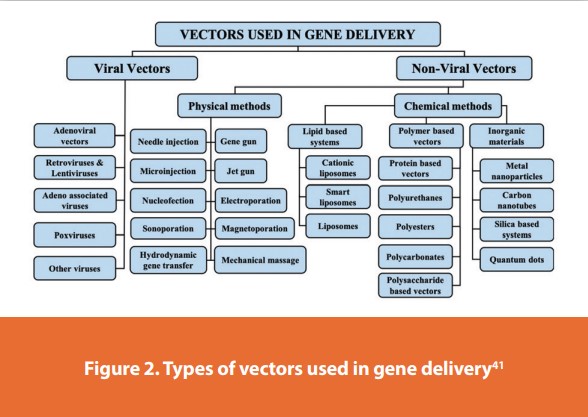
Gene therapy is the treatment of a genetic disease by the introduction of specific cell function-altering genetic material into a patient. The key step in gene therapy is efficient gene delivery to the target tissue/cells, which is carried out by gene delivery vehicles called vectors. There are two types of vectors: viral and non-viral, Figure 2.
Contemporary viral vector-based gene therapy is achieved by in vivo delivery of the therapeutic gene into the patient by vectors based on retroviruses, adenoviruses (AdVs), adeno-associated viruses (AAVs), and lentiviruses (LV). Alternatively, a therapeutic transgene can be delivered ex-vivo, whereby cells of a patient are extracted and cultured outside of the body. Cells are then genetically modified by the introduction of a therapeutic transgene and are then re-introduced back into the patient. There are four basic gene therapy approaches as follows: gene replacement, the delivery of a functional gene to replace a non-working gene; gene silencing, inactivation of a mutated gene that has become toxic to cells; gene addition, overexpression of a “foreign” or exogenous gene to impact cellular function; and gene editing, a permanent manipulation of a gene in a patient’s genome.
To ensure the process is robust, scalable, and transferable from the early phase through to commercial production; the development of producer cell lines and optimize viral vector platform for a product to simplify regulatory strategy;
- Transient transfection and plasmid system options
- Producer cell line development
- Plasmid design
- Selection of viral vector platform and cell line for vector production
- Reproducible process design with a commercialization approach
Transfection is a powerful method used to insert foreign nucleic acids into eukaryotic cells. The ability to modify host cells’ genetic content enables the broad application of this process in studying normal cellular processes, disease molecular mechanisms, and gene therapeutic effects. The characteristics, strengths, and limitations of various transfection methods, types of transfected nucleic acids, transfection controls, and approaches to assess transfection efficiency with the vast choices of approaches available.
Transfection is a process by which foreign nucleic acids are delivered into eukaryotic cells to modify the host cell’s genetic makeup.15,16 For the past 30 years, transfection has gained increasing popularity due to its wide application for studying cellular processes and molecular mechanisms of diseases.14,16,17 Understanding the molecular pathway of disease allows the discovery of specific biomarkers that may be applied to diagnose and prognose diseases.19 Transfection can be employed as one of the strategies in gene therapy to treat incurable, inherited genetic diseases.20,21,23 Today, the advancement in life-sciences technology allows different types of nucleic acids to be transfected into mammalian cells, and these include Deoxyribonucleic acids (DNAs), Ribonucleic acids (RNAs) as well as small, non-coding RNAs such as siRNA, shRNA, and miRNA.21,22,24,25
Throughout the developmental process of new therapeutics, a key factor is in the activity determination of antibody therapeutic drugs. In order to do so, transgenic cell lines are widely used due to their simplicity, speed, accuracy, non-biological safety, and animal substitution.
Activity determination could be performed in various ways, including binding studies; however, in vitro evaluation is a critical part in drug development and quality control.
Process Optimization
Optimize the viral vector development process, Figure 3, ensuring it is repeatable, scalable, and transferable to cGMP manufacturing. to ensure efficient scale-up, increase titers, reduce development timelines, and lower costs to support upstream and downstream process development. Required unit operations include;
- Research cell banks
- Master and working cell banks (MCB and MCW)
- Upstream processing
- Downstream processing
- Proof of concept evaluation runs
- Supporting processes
- Media
- Buffers
- Qualified raw materials
- Utilities
- Facility that is convertible to a cGMP operation
Comparability – ICH Q5E
Manufacturers frequently make changes to manufacturing processes during clinical development and post-approval: e.g. replacing animal-derived materials, increasing purity/consistency/yield, site transfers, and formulation changes. The goal of the comparability exercise is to ascertain that pre- and post-change drug products is comparable in terms of quality, safety, and efficacy. This is achieved via the collection and evaluation of relevant data to determine if the process change has had an adverse impact on the drug product. Data will include analytical testing, biological assays, and in some cases non-clinical and clinical data. i.e., in cases where the relationship between specific quality attributes and safety and efficacy has not been established.45
Comparability Protocol Submission
The Comparability Protocol (CP) submission should provide a comprehensive, detailed plan for the implementation of a proposed change(s) and should include the information described below. FDA uses this information to assess whether the outcomes of any proposed test or study would or would not support the specified future change(s). Such information should be sufficient to merit the proposed reduced reporting category for the implementation of the change(s).36
FDA recommends that you provide a summary of the CP submission using tabular (e.g., a list of tests and studies, analytical procedures, and acceptance criteria), narrative, and/or graphic presentations, as appropriate. The summary should include a brief overview of the following:
- A description of and rationale for the proposed CMC change(s)
- Supporting information and analysis
- Comparability protocol for the proposed change(s)
- Proposed reduced reporting category
- Other information
The questions and answers comparability considerations for ATMPs,44 Q3 states; How does the risk-based approach (RBA) apply to comparability exercises for ATMPs?
The response is; “The potential impact of the proposed change should always be evaluated for its risks to the quality of the final product and the impact on the efficacy and safety profile of the product. The overall extent of the comparability exercise for ATMPs should therefore be driven by a risk-based approach (RBA). Namely, the RBA should be used to determine an appropriate amount of comparability data and to select a suitable set of relevant critical quality attributes (CQAs) to be compared, taking into account the stage of product development and the number of batches available.
Changes that are considered to have a high risk/impact will require an extensive exercise of comparison at the in-process control level, characterization, and release. Whenever relevant, the generation of additional/new validation data has to be taken into account. On the other hand, low-risk/impact changes may entail a more limited amount of comparability data. A more comprehensive data package is required to support manufacturing changes in pivotal clinical trials or to the marketing authorization.”
Quality Risk Management – ICH Q9(R1)
Two primary principles of quality risk management (QRM):
- The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient
- The level of effort, formality, and documentation of the QRM process should be commensurate with the level of risk
ICH Q9(1) formalized quality risk assessments (QRA) and made it a proactive tool to drive continual improvement.
Determine Potential Risk of Process Change ICH Q8(R2)
Assessment of Product Critical Quality Attributes (CQAs); “A CQA is a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality. CQAs are generally associated with the drug substance, excipients, intermediates (in-process materials), and drug product.”
- CQAs are the foundation for managing product quality throughout all stages of the product lifecycle and defining the control strategy
- Assessment of the impact of the proposed process change on the defined product CQAs, based on current available process and product knowledge, allows to determine the potential risk/ impact of the change
- This will help to determine the CQAs to explore in the comparability study and the amount of data required to assess comparability, e.g. batch numbers to test, testing points in the process, additional characterization required, side-by-side testing, etc.
CQA Assessment for Proposed Process Change
Risk score guidance assessing CQAs impact due to proposed process change;
- High: Change has the potential to significantly impact CQA
- Medium: Change has the potential to minorly impact CQA
- Low: Change has no known or expected impact on CQA
Use process and product knowledge to date. If gaps in knowledge are observed: Can you do any development work beforehand to gain more knowledge? Are there other similar products you can leverage information from? Literature references? Living document, e.g. repeat the assessment once further knowledge has been gained. ICH Q5E; “The process assessment should consider such factors as the criticality of the process step and the proposed change, the location of the change and potential for effects on other process steps, and the type and extent of change”.45
Non-cGMP Vector Production
Engineering runs at variable scales for further optimization in the process development lab. Cost-effective process runs before cGMP productions. Further optimization to get to a closed and semi-automated system for vector production.
Nonclinical studies may be needed to support manufacturing changes for an investigational product after clinical studies have been initiated,13 or for a licensed product.43 If analytical studies alone are insufficient to determine the impact of the manufacturing changes on cell and gene therapy (CGT) product quality, then nonclinical studies may contribute to a demonstration of comparability.29
- Optimized cGMP runs
- Master and working cell bank productions
- Aseptic processing using closed systems from upstream processing to fill and finish
Flexible non-GMP and GMP viral vector manufacturing capacity and scale-up for both adherent and suspension platforms. Whether to adapt and develop a suspension process or scale an adherent process from research to clinical-grade production, everything should be focused on getting viral vector to commercialization.
Cell Line and Assay Testing
Accelerate the manufacturing program with analytical cell line and assay testing, by developing and qualifying a tailored process to viral vector and/or gene of interest; e.g., analytical cell line and assay testing;
- Viral reverse transcriptase assay
- Replication-competent viral assay
- Real-Time Virology Assays
- RNA titers
- Infectious units assay using flow cytometry
- P24 assay
- Host cell protein
- Host cell DNA detection
- Plasmid DNA contamination
- Endotoxin
The importance of process development (PD) for first-time-right manufacturing for clinical study and commercial readiness cannot be overstated. It is critical to increasing yields, achieving product quality expectations, and lowering the cost of goods while meeting clinical development and commercial readiness milestones.
Process Development
Process Development (PD) begins with a knowledge transfer of existing processes or the development of a custom bioprocess from scratch. PD teams then develop an optimal strategy to develop processes that generate quality material for IND-enabling studies and align equipment with future pilot-scale and cGMP-scale operations. PD and cGMP manufacturing teams need to closely collaborate and be cross-trained to ensure process performance and quality.
Define Process CQAs to Reduce Impurity Levels and Achieve Scalability
As cell and gene therapy and genomic medicine pipelines continue to mature, process development for viral vectors is becoming critical for drug developers for the next phase, to deliver a quality, transformative cure to patients with affordability. The recent investments such as Novartis-Samsung Biologics' $81m contract for viral vector manufacturing, and Resilience raising $625m towards biomanufacturing capabilities including stable cell lines for viral vector production pave the foundation for scientific and technology innovations.
With a better understanding of these novel nanoparticles, developability, and cell line development, the ability to optimize AAV (Table 1), LV formulation, and define robust CQAs are critical to drug developers’ ability to increase scalability with efficiency, reduce the cost of goods and most importantly, ensure product quality control for patients.
- Novel strategies to optimize upstream viral vector processes with improved cell line suspension
- Bridging the gap between USP and DSP, and exploring purification techniques to yield a pure viral vector drug product
- Enhance your formulation for a stable viral vector product – compare and contrast viral vector process development for cell and gene therapies
- Reduce impurity and maximize process development robustness for greater process control and quality
- Navigate the fast-evolving regulatory landscape to achieve GMP during scale-up and scale-out
Considerations for the Management of Changes
An effective quality system maintains consistency in drug product (DP) quality throughout the product lifecycle, including by adequately managing manufacturing changes. In general, manufacturing changes should be thoroughly assessed and documented using effective change control procedures. For investigational products, maintaining product quality by control of CQAs and critical process parameters (CPPs) during manufacturing changes is important for obtaining interpretable clinical study data that can support licensure. A robust framework for managing manufacturing changes is especially valuable for CGT products because of the complexity of these products and their manufacturing processes.
The management of manufacturing changes presents many challenges for human CGT products due to the complexity of these products.13
FDA provides sponsors of Investigational New Drug Applications (INDs) and applicants of Biologics License Applications (BLAs) for CGT products, with recommendations regarding product comparability and the management of manufacturing changes for investigational and licensed CGT products.
CGT products are regulated under the existing framework for biological products. Manufacturing and control of CGT products can often be affected by unique factors, including limited knowledge of product quality attributes, limited manufacturing experience, limited and variable starting materials, limited amount of product, complex manufacturing processes, and limited product shelf life. These aspects of CGT products may make the management of manufacturing changes more challenging than for other biological products.
A CGT product manufacturer may seek to implement a manufacturing change for a variety of reasons, including improving product quality, expanding product supply, or improving manufacturing efficiency. The risk that a manufacturing change may adversely impact product quality should be prospectively assessed under the manufacturer’s quality risk management processes.27,28 It is noted that while improvement of product quality is always desirable and encouraged, if the results of comparability studies indicate an improved product quality suggesting a significant benefit in effectiveness and/or safety, the pre-and post-change products may be different products and, therefore, not comparable.
Risk assessment should be performed for all types of manufacturing changes, regardless of the stage of product development. If a risk assessment indicates that a manufacturing change has the potential to adversely affect product quality, comparability studies should be performed to evaluate the impact of the proposed manufacturing change. It can be difficult to fully characterize CGT products using analytical methods, and in some cases, analytical studies alone may not be sufficient to reach a conclusion regarding comparability. In such cases, additional data from nonclinical studies may help to support comparability. Otherwise, additional clinical studies may be warranted.
CMC Changes Requiring New IND Submission
Some changes can fundamentally alter the design or nature of the product, resulting in a new product. Initiation of clinical studies with the new investigational product generally requires the submission of a separate IND (21 CFR 312.20). It is recommended that you seek FDA advice (section VII of this guidance: Draft Guidance for Industry, July 2023; Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products; Draft Guidance for Industry) regarding any manufacturing changes that could alter the product and require a new IND. Some examples of changes that may require a new IND include:26
- Change in the cellular starting material of a cellular product (e.g., allogeneic vs. autologous donor; adipose-derived cells vs. umbilical cord-derived cells)
- Change to the types of cells in a cellular product (e.g., a mixture of CD4+ and CD8+ T cells instead of solely CD4+ T cells)
- Change to the scaffold or matrix component of the final construct in tissue-engineered medical products (TEMPs) (e.g., changes to chemical or physical properties) causing significant modification to the product characteristics
- Change in a viral vector capsid or envelope that changes the tropism or serotype of a viral vector used for in vivo gene therapy
- Change to the sequence of a transgene or addition of a transgene (e.g., changes to the intracellular signaling domain of a chimeric antigen receptor)
- Change in expression control elements of a viral vector (e.g., change from a tissue-specific to a ubiquitous promoter)
- Change of target gene for genome editing products, including the addition of a target gene
Reporting Manufacturing Changes to a BLA
For licensed products, you must report each change in the product, production process, quality controls, equipment, facilities, responsible personnel, or labeling established in the approved license application, by the requirements in 21 CFR 601.12. When reporting these changes, your supplement or annual report should include a risk assessment report and must include data from appropriate studies performed to evaluate the effect of the changes on product quality as required under 21CFR 601.12.43,90
To facilitate the management of post-approval manufacturing changes, you may submit one or more comparability protocols to your BLA for FDA review, as described in Licensing, Changes to an approved application. eCFR:21 CFR 601.12 -- Changes to an approved application. These protocols may be submitted either in the original BLA or, if the application is already approved, in a prior approval supplement. Comparability protocols should be located in section 3.2.R of the BLA. Upon approval, this protocol becomes an agreed-upon plan for implementation of the manufacturing change using the reporting category specified in the approved comparability protocol submitted under43. provided that there is successful completion of the plan for implementation of the change(s) as described in the comparability protocol, including achievement of all of the predefined acceptance criteria for success in the approved comparability protocol.36
Risk Assessment
Manufacturing changes that can present potential risks to product quality include, but are not limited to, changes to the manufacturing site, manufacturing process, materials, container closure, testing, storage, and shipping conditions. To evaluate whether the proposed manufacturing change may impact product quality, you should conduct a detailed risk assessment as recommended in International Council for Harmonization (ICH) Q9(R1) dated July 2023.27 The process of evaluating the risk of a manufacturing change for a CGT product is similar to risk evaluation for other types of drugs, and the same tools can generally be applied.
We recognize that risk assessment for changes to the manufacturing of CGT products may be more challenging than for other product types because the effects of manufacturing changes are often difficult to predict for these complex products. For example, manufacturing changes may unexpectedly alter product purity (increase process-related impurities, cellular impurities, aggregates, or particulates), reduce product stability, or change product potency.
Transferring a manufacturing process to a new manufacturing facility is generally considered a major change that may require extensive comparability evaluation in addition to technology transfer, because it may involve changes to the manufacturing process, shipping, manufacturing equipment, testing equipment, and operators. Performing a thorough risk assessment, including consideration of method equivalence and CPPs, is essential when transferring a manufacturing process to a new facility.
Your risk assessment should consider the potential impacts of the change on the manufacturing steps and in-process parameters that are downstream of the manufacturing change, as well as the impact on the product. It is recommended that you take a stepwise approach to select all quality attributes and process parameters to be evaluated in a comparability study; first, you should determine which attributes might be affected by the particular change, and then you should assign a score to each attribute based on the probability, severity, and detectability of the risk. The assigned score can be used to determine the overall risk for each attribute. Manufacturing changes that are determined to have a high risk to product quality should be supported by an extensive analytical comparability study, while it may be possible to evaluate low-risk changes using a more focused approach.26
You should consider whether your risk assessment is constrained by gaps in product knowledge related to the type of change being proposed. Gaps in knowledge typically raise the level of risk and may necessitate a more extensive comparability study. Note that relying solely on established release tests and in-process controls is generally insufficient to assess the impact of manufacturing changes. Therefore, it is recommended that you consider the potential impact of manufacturing changes on quality attributes that are not routinely evaluated by established release tests and process controls, and consider additional characterization studies as appropriate. Additionally, your risk assessment should evaluate whether more than one analytical method should be used to evaluate a particular attribute. Such an approach could be useful for high-risk attributes, particularly with respect to the assessment of potency, as described in section V.B (Analytical Comparability Study Design) of this guidance.26 In your risk assessment, you should justify how the selected quality attributes and process parameters can be used to comprehensively evaluate the potential effect of the change on product quality.
Your risk assessment should also inform the statistical approach to comparability. Higher-risk attributes typically warrant a more stringent statistical analysis than lower-risk attributes. Side-by-side or graphical presentations (such as dot plots) to allow visual comparison, in lieu of statistical analysis, may be sufficient for the characterization of attributes at low risk of being impacted by a manufacturing change.
It is important to note that a manufacturing change may affect product stability even if the change has no other effect on product quality or process performance. As discussed in section III.B,26 you should assess the potential risk to product stability and delivery device compatibility.
Finally, if multiple changes are to be implemented simultaneously, it is recommended that you assess the risk of each individual change and the potential cumulative effect of the changes on product quality. It may be possible to evaluate these multiple changes under a single comparability study. However, if you fail to demonstrate comparability in this single study, it will likely be difficult to identify which of the changes caused an adverse effect on product quality.26
Phase-Dependent Approaches to Plasmid DNA Manufacture
Plasmid DNA lies at the heart of viral and mRNA vector production because it provides the coding sequences for gene-based advanced therapy medicinal products (ATMPs). Its manufacture is therefore critical to both the supply and success of these products.
The manufacture of plasmid DNA is certainly critical if viral vectors are produced via transient transfection. This production route calls for the manufacture of up to four plasmids. Otherwise, production may not lead to an initial vector suitable for first-in-human studies.
Production routes that require multiple plasmids put additional pressure on plasmid manufacture, which also needs to supply enough plasmids to sustain growth in the ATMP market. This growth is evident in the number of products in development, as well as in the number of products entering late clinical phase or commercial production. If this growth is to continue, plasmid suppliers will need to modify their service offerings.
The market for plasmid DNA is unusual in that its customers—vector developers—straddle two somewhat conflicting perspectives. That is, these developers see plasmids as commodity products that can be sourced from multiple suppliers. However, the developers also recognize that plasmids pose regulatory challenges because plasmid suppliers maintain their own manufacturing processes and, more critically, their own approaches to quality and testing. Vector developers need to reconcile these perspectives not just during the production and procurement of plasmid DNA, but also through subsequent stages of clinical development.
Plasmid Production Platforms
Plasmid producers have their own platform processes for the manufacture of plasmid DNA. These processes may be used for multiple plasmid products without being extensively optimized. Although various processes are in use, they all follow the general outline shown in Figure 4.
Processes are based on high-density fermentation, usually in a K12-based Escherichia coli strain, the aim being to achieve high specific productivities by constraining growth rates and minimizing the generation of open and linear plasmid forms during the fermentation.
In terms of downstream processing, the key purification operations are the cell lysis steps46 and the chromatography steps (which remove key host contaminants and, critically, separate open and closed plasmid forms). A vital element of these processes is their robustness in handling different plasmids, including plasmids of significantly different sizes. (Sizes may range from 2 kb to 16 kb.) These processes also seek to sustain productivity, limiting the need for development process studies and, importantly, keeping timelines short and containing costs.
Traditionally, plasmids were produced in fixed systems. However, with the increasing need to achieve complete product segregation, the drive is toward production in single-use systems. This presents its own challenges around fermentation, access to equipment, and other key process steps. Although production scales vary from milligram-scale to hundred-gram-scale processes, most systems remain below the 1,000-L scale.
Regulatory
Another plasmid manufacturing issue is quality assurance. Devising a suitable quality assurance system can be challenging because plasmids are seldom final drug substances. In fact, plasmids may be several steps removed from final drug substances.
When vector production began, there were limited guidelines on the quality of plasmid to be used, and producers took very different approaches. To summarize, in 2005 the European Medicines Agency (EMA) produced guidelines for the production of lentiviral vectors, which required that the plasmid be of a “high quality.” However, the EMA did not define the standards that a plasmid should meet. More recently, in 2018, the EMA issued updated guidelines in which plasmid was designated as a starting material. Consequently, it became necessary to sequence and identify plasmids. In 2020, the U.S. Food and Drug Administration (FDA) disseminated recommendations regarding chemistry, manufacturing, and control information submitted in investigational new drug applications. In 2023, the EMA issued more extensive guidance around the grades of plasmid required for the production of a range of ATMPs.
The guidance provides examples of where Good Manufacturing Practice (GMP) regulations or GMP principles apply in the manufacturing process for ATMPs. It also offers suggestions for the use of risk-based approaches around plasmid supply and identifies the need for technical agreements with plasmid suppliers.
Although this document does not propose a requirement for GMP-grade plasmid or Qualified Person release of plasmid, it does suggest the need for formal technical agreements, placing the onus on vector producers to take a risk-based approach around plasmid supply during clinical development and, subsequently, during commercial supply. Progression of cGMP compliance at different stages of product development; Illustration of the increase in the level of compliance with cGMP as products progress from a pre-clinical stage to the Biological License Application and Commercial Manufacturing, Figure 5.
The production of plasmid DNA is becoming an increasingly important element in the supply chain for ATMPs. It is therefore going to be evermore critical that plasmid suppliers work with vector producers to ensure that plasmid supplies are maintained while achieving the required quality standards through the development and commercialization of these potentially life-saving medicines. Plasmid suppliers and vector producers can work together effectively if they follow risk-based and phase-dependent approaches.
Viral Vector Production
Viral vector production employs complex processes resulting in varying challenges throughout the product lifecycle. The main issues revolve around selecting a production system, optimizing product quality, and building standardization to enable a robust CMC approach.
With the discovery of new fields of application for viral vectors in gene therapy comes an increased need for dedicated solutions in manufacturing and supply chains. A wide product portfolio range of manufacturers’ demands:
- Primary packaging (suitable for cryopreservation)
- Fluid Management Solutions
- Cryogenic Freezers
- Ultra-Cold Storage Freezers
- Shipping Containers
Together, these products comprise an elaborate product array designed to facilitate the medical breakthroughs enabled by viral vectors.
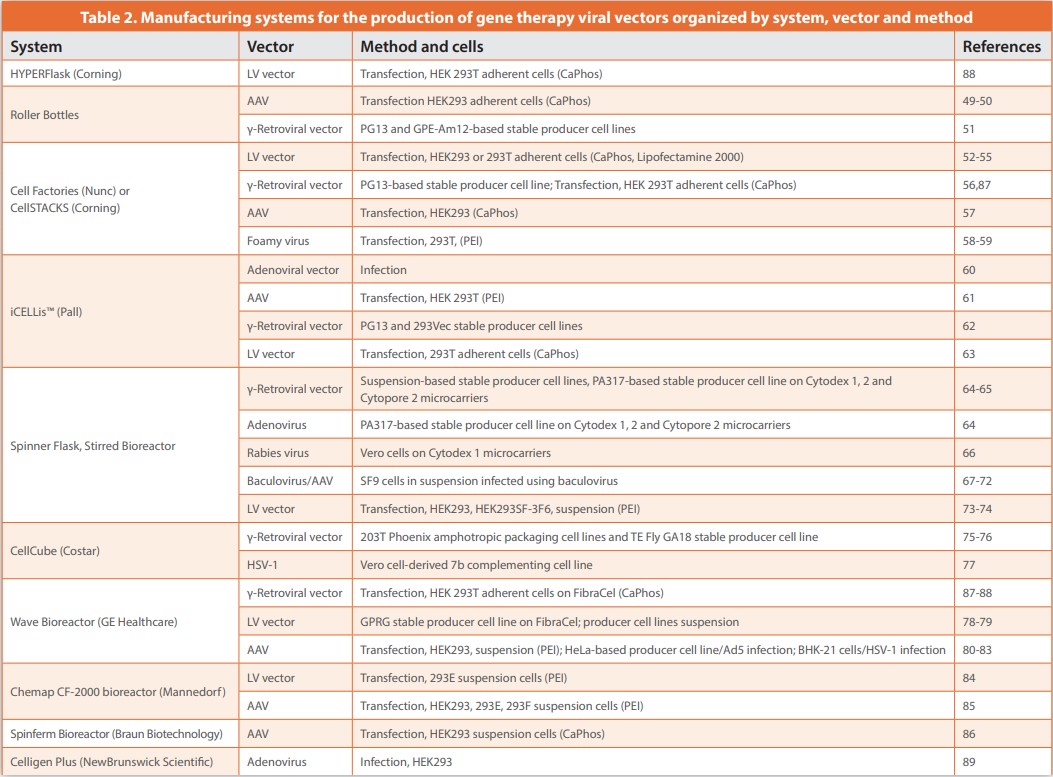
Manufacturing processes for viral vectors include a variety of approaches, predominantly based on the use of mammalian cells in either adherent or suspension-cell-based systems. Traditional laboratory-scale systems with adherent cells are generally difficult to scale up due to the large number of flasks, roller bottles, or cell factories that need to be manipulated during a clinical production run. Not only does this pose a challenge in terms of available incubator space, but the manipulation of a large number of culture vessels increases processing time and risk due to the number of steps that include open manipulation during aseptic processing. To address this challenge, and to reduce risk during clinical manufacturing, the field is moving to larger single-use disposable culture systems and bioreactors, Figure 6. A variety of different systems have been used, mostly defined by the type of vector to be manufactured and the type of cells used, with examples of each provided in Table 2. The choice of system is also affected by whether or not the virus is harvested from the cell culture media, from the cells, or from both.
References
- Edelstein M. The Journal of Gene Medicine Clinical Trial site. Gene Therapy Clinical Trials Worldwide. http://www.wiley.com//legacy/wileychi/genmed/clinical/ (October 2015, date last accessed).
- Herper M. (2014) Gene Therapy’s Big Comeback. In: Forbes, April 14. http://www.forbes. com/sites/matthewherper/2014/03/26/once-seen-as-too-scary-editing-peoples-genes-with-viruses-makes-a-618-million-comeback/ (October 2015, date last accessed). [Google Scholar]
- Philippidis A. (2014) Gene therapy briefs. Hum. Gene. Ther., 25, 262–264. [PubMed] [Google Scholar]
- Chaudhary G. (2015) Gene therapy market to be worth over USD 10 Billion by 2025, Predicts Roots Analysis. Business Wire, London. March 3, 2015.
- Roots Analysis. (2015) Gene therapy market, 2015–2025.
- Yla-Herttuala S. (2012) Endgame: glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol. Ther., 20, 1831–1832. [PMC free article] [PubMed] [Google Scholar]
- Peng Z. (2005) Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene. Ther., 16, 1016–1027. [PubMed] [Google Scholar]
- Liang M. (2012) Clinical development of oncolytic viruses in China. Curr. Pharm. Biotechnol., 13, 1852–1857. [PubMed] [Google Scholar]
- Gordon E.M., Hall F.L. (2010) Rexin-G, a targeted genetic medicine for cancer. Expert. Opin. Biol. Ther., 10, 819–832. [PubMed] [Google Scholar]
- Human Stem Cells Institute (2011) HSCI receives approval to market neovasculgen – the first Russian gene-therapy drug for the treatment of peripheral arterial disease. Human Stem Cells Institute, Moscow, Russia. December 7, 2011.
- Spark Therapeutics (2015) Spark therapeutics announces positive top-line results from pivotal Phase 3 Trial of SPK-RPE65 for genetic blinding conditions. Globe Newswire, Philadelphia, PA. October 5, 2015.
- Schimmer J., Breazzano S. (2015) Investor outlook: focus on upcoming LCA2 gene therapy Phase III results. Hum. Gene. Ther. Clin. Dev., 26, 144–149. [PubMed] [Google Scholar]
- Huggett B. (2014) Innovative startups 2013. Nat. Biotechnol., 32, 127. [PubMed] [Google Scholar]
- Arnold et al. (2006) Arnold AS, Laporte V, Dumont S, Appert-Collin A, Erbacher P, Coupin G, Levy R, Poindron P, Gies JP. Comparing reagents for efficient transfection of human primary myoblasts: FuGENE 6, effectene and ExGen 500. Fundamental and Clinical Pharmacology. 2006;20(1):81–89. doi: 10.1111/j.1472-8206.2005.00344.x. [PubMed] [CrossRef] [Google Scholar]
- Kim & Eberwine (2010) Kim TK, Eberwine JH. Mammalian cell transfection: the present and the future. Analytical and Bioanalytical Chemistry. 2010;397(8):3173–3178. doi: 10.1007/ s00216-010-3821-6. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Chow et al. (2016) Chow YT, Chen S, Wang R, Liu C, Kong C-W, Li RA, Cheng SH, Sun D. Single cell transfection through precise microinjection with quantitatively controlled injection volumes. Scientific Reports. 2016;6(1):24127. doi: 10.1038/srep24127. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Ishida & Selaru (2012) Ishida M, Selaru FM. miRNA-based therapeutic strategies. Current Anesthesiology Reports. 2012;1(1):63–70. doi: 10.1007/s40139-012-0004-5. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- J. Wright ‘Quality Control Testing, Characterization and Critical Quality Attributes of Adeno-Associated Virus Vector Used for Human Gene Therapy’, Biotechnol J. 2021 Jan;16(1)
- Roser et al. (2018) Roser AE, Gomes LC, Schünemann J, Maass F, Lingor P. Circulating miRNAs as diagnostic biomarkers for Parkinson’s disease. Frontiers in Neuroscience. 2018;12:625. doi: 10.3389/fnins.2018.00625. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Yao et al. (2008) Yao CP, Zhang ZX, Rahmanzadeh R, Huettmann G. Laser-based gene transfection and gene therapy. IEEE Transactions on Nanobioscience. 2008;7(2):111–119. doi: 10.1109/TNB.2008.2000742. [PubMed] [CrossRef] [Google Scholar]
- Tomizawa et al. (2013) Tomizawa M, Shinozaki F, Motoyoshi Y, Sugiyama T, Yamamoto S, Sueishi M. Sonoporation: gene transfer using ultrasound. World Journal of Methodology. 2013;3(4):39–44. doi: 10.5662/wjm.v3.i4.39. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Borawski et al. (2007) Borawski J, Lindeman A, Buxton F, Labow M, Gaither LA. Optimization procedure for small interfering RNA transfection in a 384-well format. Journal of Biomolecular Screening. 2007;12(4):546–559. doi: 10.1177/1087057107300172. [PubMed] [CrossRef] [Google Scholar]
- Yamano, Dai & Moursi (2010) Yamano S, Dai J, Moursi AM. Comparison of transfection efficiency of nonviral gene transfer reagents. Molecular Biotechnology. 2010;46(3):287– 300. doi: 10.1007/s12033-010-9302-5. [PubMed] [CrossRef] [Google Scholar]
- Sork et al. (2016) Sork H, Nordin JZ, Turunen JJ, Wiklander OP, Bestas B, Zaghloul EM, Margus H, Padari K, Duru AD, Corso G, Bost J, Vader P, Pooga M, Smith CE, Wood MJ, Schiffelers RM, Hällbrink M, Andaloussi SEL. Lipid-based transfection reagents exhibit cryo-induced increase in transfection efficiency. Molecular Therapy—Nucleic Acids. 2016;5(3):e290. doi: 10.1038/mtna.2016.8. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Shi et al. (2018) Shi B, Xue M, Wang Y, Wang Y, Li D, Zhao X, Li X. An improved method for increasing the efficiency of gene transfection and transduction. International Journal of Physiology, Pathophysiology and Pharmacology. 2018;10(2):95–104. [PMC free article] [PubMed] [Google Scholar]
- Guidance Document, manufacturing changes and comparability for human cellular and gene therapy products, Draft Guidance for Industry, July 2023; Manufacturing Changes and Comparability for Human Cellular and Gene Therapy Products; Draft Guidance for Industry (fda.gov)
- Guidance for Industry: Q9(R1) Quality Risk Management, 26 July 2023, ICH guideline Q9 (R1) on quality risk management (europa.eu).
- Guidance for Industry: Q10 Pharmaceutical Quality System, April 2009, https://www.fda. gov/media/71553/download.
- Guidance for Industry: Q8(R2) Pharmaceutical Development, November 2009, https:// www.fda.gov/media/71535/download.
- Guidance for Industry: Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology-derived Products, April 1996, https://www.fda.gov/ regulatory-information/search-fda-guidancedocuments/demonstration-comparability-human-biological-products-includingtherapeutic-biotechnology-derived.
- Guidance for Industry: Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process, June 2005, https://www.fda.gov/ media/71489/download.
- Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products; Guidance for Industry, June 2021, https://www.fda.gov/ media/109615/download.
- Guidance for Industry: Process Validation: General Principles and Practices, January 2011, https://www.fda.gov/media/71021/download.
- Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products, November 2013, https://www.fda.gov/media/87564/download.
- Guidance for Industry: M4Q: The CTD – Quality, August 2001, https://www.fda.gov/ media/71581/download.
- FDA Guidance for Industry: Comparability Protocols for Post-approval Changes to the Chemistry, Manufacturing, and Controls Information in an NDA, ANDA, or BLA, October 2022, https://www.fda.gov/media/162263/download.
- Guidance for Industry: CGMP for Phase I Investigational Drugs, July 2008, https://www. fda.gov/media/70975/download.
- Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products, January 2011, https://www.fda.gov/media/79856/download.
- Analytical Procedures and Methods Validation for Drugs and Biologics; Guidance for Industry, July 2015, https://www.fda.gov/media/87801/download.
- Q2(R1) Validation of Analytical Procedures: Text and Methodology: Guidance for Industry, September 2021, https://www.fda.gov/media/152208/download.
- Muhammad Hammad Butt, Muhammad Zaman, Abrar Ahmad, Rahima Khan, Tauqeer Hussain Mallhi, Mohammad Mehedi Hasan, Yusra Habib Khan, Sara Hafeez, Ehab El Sayed Massoud, Md Habibur Rahman, Simona Cavalu; “Appraisal for the Potential of Viral and Nonviral Vectors in Gene Therapy: A Review”, July 30, 2022. PMID: 36011281, PMCID: PMC9407213, DOI: 10.3390/genes13081370
- Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products, November 2013, https://www.fda.gov/media/87564/download
- Title 21, Food and Drugs, Chapter I-Food and Drug Administration, Department of Health and Human Services, Part 601.12 – Licensing, Changes to an approved application. eCFR :: 21 CFR 601.12 -- Changes to an approved application.
- Questions and answers; Comparability considerations for Advanced Therapy Medicinal Products (ATMPs), (EMA/CAT/499821/2019). https://www.ema.europa.eu/en/ documents/other/questions-and-answers-comparability-considerations-advanced-therapy-medicinal-products-atmp_en.pdf
- Q5E Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process, June 2005. FDA-2004-D-0440. https://www.fda. gov/regulatory-information/search-fda-guidance-documents/q5e-comparability[1]biotechnologicalbiological-products-subject-changes-their-manufacturing-process
- Birnboim H. C., Doly J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7, 1513–1523. [PMC free article] [PubMed] [Google Scholar]
- On October 12, 2017, an FDA Advisory Committee voted 16 to 0 in recommending marketing approval for LUXTURNA; History Is Made: FDA Approves Spark’s Vision-Restoring Gene Therapy — Foundation Fighting Blindness
- Kutner R.H., Puthli S., Marino M.P., Reiser J. (2009) Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC. Biotechnol., 9, 10. [PMC free article] [PubMed] [Google Scholar]
- Wright J.F., Wellman J., High K.A. (2010) Manufacturing and regulatory strategies for clinical AAV2-hRPE65. Curr. Gene. Ther., 10, 341–349. [PubMed] [Google Scholar]
- Liu Y.L., Wagner K., Robinson N., Sabatino D., Margaritis P., Xiao W., Herzog R.W. (2003) Optimized production of high-titer recombinant adeno-associated virus in roller bottles. Biotechniques, 34, 184–189. [PubMed] [Google Scholar]
- Cornetta K., Matheson L., Ballas C. (2005) Retroviral vector production in the National Gene Vector Laboratory at Indiana University. Gene. Ther., 12(Suppl 1), S28–S35. [PubMed] [Google Scholar]
- Geraerts M., Michiels M., Baekelandt V., Debyser Z., Gijsbers R. (2005) Upscaling of lentiviral vector production by tangential flow filtration. J. Gene. Med., 7, 1299–1310. [PubMed] [Google Scholar]
- Karolewski B.A., Watson D.J., Parente M.K., Wolfe J.H. (2003) Comparison of transfection conditions for a lentivirus vector produced in large volumes. Hum. Gene. Ther., 14, 1287– 1296. [PubMed] [Google Scholar]
- Merten O.W., Charrier S., Laroudie N., Fauchille S., Dugue C., Jenny C., Audit M., Zanta-Boussif M.A., Chautard H., Radrizzani M. et al. (2011) Large-scale manufacture and characterization of a lentiviral vector produced for clinical ex vivo gene therapy application. Hum. Gene. Ther., 22, 343–356. [PubMed] [Google Scholar]
- Slepushkin V., Chang N., Cohen R., Gan Y., Jiang B., Deausen E., VBerlinger D., Binder G., Andre K., Hummeau L. et al. (2003) Large-scale purification of a lentiviral vector by size exclusion chromatography or mustang Q ion exchange capsule. Bioprocessing. J., 2, 89–95. [Google Scholar]
- Przybylowski M., Hakakha A., Stefanski J., Hodges J., Sadelain M., Riviere I. (2006) Production scale-up and validation of packaging cell clearance of clinical-grade retroviral vector stocks produced in cell factories. Gene. Ther., 13, 95–100. [PubMed] [Google Scholar]
- Lock M., Alvira M., Vandenberghe L.H., Samanta A., Toelen J., Debyser Z., Wilson J.M. (2010) Rapid, simple, and versatile manufacturing of recombinant adeno-associated viral vectors at scale. Hum. Gene. Ther., 21, 1259–1271. [PMC free article] [PubMed] [Google Scholar]
- Nasimuzzaman M., Lynn D., Beuerlein M., Cross S., Link K., Lutzko C.M., Nordling D.L., Russell D.W., Malik P., van der Loo J.C.M. (2015) Scale-up and manufacturing of high-titer foamy virus vector containing human CD18 for the treatment of leukocyte adhesion deficiency. Poster 461, In American Society of Gene & Cell Therapy, New Orleans, LA, May 13–16, 2015. [Google Scholar]
- Nasimuzzaman M., Kim Y.S., Wang Y.D., Persons D.A. (2014) High-titer foamy virus vector transduction and integration sites of human CD34(+) cell-derived SCID-repopulating cells. Mol. Ther. Methods. Clin. Dev., 1, 14020. [PMC free article] [PubMed] [Google Scholar]
- Lesch H.P., Heikkila K.M., Lipponen E.M., Valonen P., Muller A., Rasanen E., Tuunanen T., Hassinen M.M., Parker N., Karhinen M. et al. (2015) Process development of adenoviral vector production in fixed bed bioreactor: from bench to commercial scale. Hum. Gene. Ther., 26, 560–571. [PubMed] [Google Scholar]
- Emmerling V.V., Pegel A., Milian E.G., Venereo-Sanchez A., Kunz M., Wegele J., Kamen A.A., Kochanek S., Hoerer M. (2015) Rational plasmid design and bioprocess optimization to enhance recombinant adeno-associated virus (AAV) productivity in mammalian cells. Biotechnol. J., 10.1002/biot.201500176. [PubMed] [CrossRef] [Google Scholar]
- Wang X., Olszewska M., Qu J., Wasielewska T., Bartido S., Hermetet G., Sadelain M., Riviere I. (2015) Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J. Immunother., 38, 127–135. [PMC free article] [PubMed] [Google Scholar]
- Valkama A.J., Lesch H.P., Martikainen A., Malinen J., Salonen T., Mahonen M., Heikura T., Yla-Herttuala S. (2015) Optimization of lentiviral vector production in a fixed-bed bioreactor. In European Society of Gene and Cell Therapy, Vol. P155. [PMC free article] [PubMed]
- Wu S.C., Huang G.Y., Liu J.H. (2002) Production of retrovirus and adenovirus vectors for gene therapy: a comparative study using microcarrier and stationary cell culture. Biotechnol. Prog., 18, 617–622. [PubMed] [Google Scholar]
- Pizzato M., Merten O.W., Blair E.D., Takeuchi Y. (2001) Development of a suspension packaging cell line for production of high titre, serum-resistant murine leukemia virus vectors. Gene. Ther., 8, 737–745. [PubMed] [Google Scholar]
- Rourou S., van der Ark A., Majoul S., Trabelsi K., van der Velden T., Kallel H. (2009) A novel animal-component-free medium for rabies virus production in Vero cells grown on Cytodex 1 microcarriers in a stirred bioreactor. Appl. Microbiol. Biotechnol., 85, 53–63. [PubMed] [Google Scholar]
- Virag T., Cecchini S., Kotin R.M. (2009) Producing recombinant adeno-associated virus in foster cells: overcoming production limitations using a baculovirus-insect cell expression strategy. Hum. Gene. Ther., 20, 807–817. [PMC free article] [PubMed] [Google Scholar]
- Cecchini S., Negrete A., Kotin R.M. (2008) Toward exascale production of recombinant adeno-associated virus for gene transfer applications. Gene. Ther., 15, 823–830. [PubMed] [Google Scholar]
- Cecchini S., Virag T., Kotin R.M. (2011) Reproducible high yields of recombinant adeno-associated virus produced using invertebrate cells in 0.02- to 200-liter cultures. Hum. Gene. Ther., 22, 1021–1030. [PMC free article] [PubMed] [Google Scholar]
- Urabe M., Ding C., Kotin R.M. (2002) Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene. Ther., 13, 1935–1943. [PubMed] [Google Scholar]
- Negrete A., Kotin R.M. (2007) Production of recombinant adeno-associated vectors using two bioreactor configurations at different scales. J. Virol. Methods, 145, 155–161. [PMC free article] [PubMed] [Google Scholar]
- Kotin R.M. (2011) Large-scale recombinant adeno-associated virus production. Hum. Mol. Genet., 20, R2–R6. [PMC free article] [PubMed] [Google Scholar]
- Segura M.M., Garnier A., Durocher Y., Ansorge S., Kamen A. (2010) New protocol for lentiviral vector mass production. Methods. Mol. Biol., 614, 39–52. [PubMed] [Google Scholar]
- Ansorge S., Lanthier S., Transfiguracion J., Durocher Y., Henry O., Kamen A. (2009) Development of a scalable process for high-yield lentiviral vector production by transient transfection of HEK293 suspension cultures. J. Gene. Med., 11, 868–876. [PubMed] [Google Scholar]
- Merten O.W., Cruz P.E., Rochette C., Geny-Fiamma C., Bouquet C., Goncalves D., Danos O., Carrondo M.J. (2001) Comparison of different bioreactor systems for the production of high titer retroviral vectors. Biotechnol. Prog., 17, 326–335. [PubMed] [Google Scholar]
- Wikstrom K., Blomberg P., Islam K.B. (2004) Clinical grade vector production: analysis of yield, stability, and storage of gmp-produced retroviral vectors for gene therapy. Biotechnol. Prog., 20, 1198–1203. [PubMed] [Google Scholar]
- Ozuer A., Wechuck J.B., Goins W.F., Wolfe D., Glorioso J.C., Ataai M.M. (2002) Effect of genetic background and culture conditions on the production of herpesvirus-based gene therapy vectors. Biotechnol. Bioeng., 77, 685–692. [PubMed] [Google Scholar]
- Palfi S., Gurruchaga J.M., Ralph G.S., Lepetit H., Lavisse S., Buttery P.C., Watts C., Miskin J., Kelleher M., Deeley S. et al. (2014) Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: a dose escalation, open-label, phase 1/2 trial. Lancet, 383, 1138–1146. [PubMed] [Google Scholar]
- Greene M.R., Lockey T., Mehta P.K., Kim Y.S., Eldridge P.W., Gray J.T., Sorrentino B.P. (2012) Transduction of human CD34+ repopulating cells with a self-inactivating lentiviral vector for SCID-X1 produced at clinical scale by a stable cell line. Hum. Gene. Ther. Methods., 23, 297–308. [PMC free article] [PubMed] [Google Scholar]
- Grieger J.C., Soltys S.M., Samulski R.J. (2015) Production of recombinant adeno-associated virus vectors using suspension HEK293 cells and continuous harvest of vector from the culture media for GMP FIX and FLT1 clinical vector. Mol. Ther., 10.1038/mt.2015.187. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Thorne B.A., Takeya R.K., Peluso R.W. (2009) Manufacturing recombinant adeno-associated viral vectors from producer cell clones. Hum. Gene. Ther., 20, 707–714. [PubMed] [Google Scholar]
- Farson D., Harding T.C., Tao L., Liu J., Powell S., Vimal V., Yendluri S., Koprivnikar K., Ho K., Twitty C. et al. (2004) Development and characterization of a cell line for large-scale, serum-free production of recombinant adeno-associated viral vectors. J. Gene. Med., 6, 1369–1381. [PubMed] [Google Scholar]
- Thomas D.L., Wang L., Niamke J., Liu J., Kang W., Scotti M.M., Ye G.J., Veres G., Knop D.R. (2009) Scalable recombinant adeno-associated virus production using recombinant herpes simplex virus type 1 coinfection of suspension-adapted mammalian cells. Hum. Gene. Ther., 20, 861–870. [PubMed] [Google Scholar]
- Segura M.M., Garnier A., Durocher Y., Coelho H., Kamen A. (2007) Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng., 98, 789–799. [PubMed] [Google Scholar]
- Durocher Y., Pham P.L., St-Laurent G., Jacob D., Cass B., Chahal P., Lau C.J., Nalbantoglu J., Kamen A. (2007) Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J. Virol. Methods., 144, 32–40. [PubMed] [Google Scholar]
- Park J.Y., Lim B.P., Lee K., Kim Y.G., Jo E.C. (2006) Scalable production of adeno-associated virus type 2 vectors via suspension transfection. Biotechnol. Bioeng., 94, 416–430. [PubMed] [Google Scholar]
- van der Loo J.C., Swaney W.P., Grassman E., Terwilliger A., Higashimoto T., Schambach A., Baum C., Thrasher A.J., Williams D.A., Nordling D.L. et al. (2012) Scale-up and manufacturing of clinical-grade self-inactivating gamma-retroviral vectors by transient transfection. Gene. Ther., 19, 246–254. [PMC free article] [PubMed] [Google Scholar]
- van der Loo J.C., Swaney W.P., Grassman E., Terwilliger A., Higashimoto T., Schambach A., Hacein-Bey-Abina S., Nordling D.L., Cavazzana-Calvo M., Thrasher A.J. et al. (2012) Critical variables affecting clinical-grade production of the self-inactivating gamma-retroviral vector for the treatment of X-linked severe combined immunodeficiency. Gene. Ther., 19, 872–876. [PMC free article] [PubMed] [Google Scholar]
- Peng Z. (2005) Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum. Gene. Ther., 16, 1016–1027. [PubMed] [Google Scholar]
- Chemistry, Manufacturing, and Controls Changes to an Approved Application: Certain Biological Products; Guidance for Industry, June 2021, https://www.fda.gov/ media/109615/download.
Author Details
Robert Dream- Managing Director, HDR COMPANY LLC
Publication Details
This article appeared in American Pharmaceutical Review:Vol. 27, No. 3April 2024Pages: 34-46
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!