Introduction
Lipid based delivery systems continue to attract considerable interest within the pharmaceutical arena, not only because of their ability to improve bioavailability via their interaction with the components of the gastrointestinal tract [1-5] but also because they may be used to manipulate release rates in vitro and in vivo [6-10]. One of the challenges associated with their use, however, is that their consistency tends to be liquid, semi-solid or, at best, a relatively pliable solid, which does not lend itself well to conventional dosage form preparation techniques. In mitigation, however, they do tend to melt at low temperatures and in a reversible manner without degradation, hence such systems are suitable for techniques whereby liquefaction is an inherent step.
In this article, we focus on the use of spray chilling as a means of preparing lipid-based dosage forms. This technique is generating considerable interest as a relatively inexpensive, simple and versatile means of particle generation while also being extremely well suited to lipid systems for the reasons outlined above. We illustrate these principles with reference to two dosage form systems, which relate to the issues of taste masking and improved bioavailability.
Spray Chilling as a Pharmaceutical Process
 The basic principles of spray chilling are, as stated above extremely simple, although inevitably there are numerous opportunities and subtleties associated with optimizing the process; the interested reader is referred to a number of sources, which discuss the processing aspects in more detail [11-19]. In brief, the technique involves the mixing of drug and lipid in a heated pressurized container such that the system is brought into the molten state. The resulting liquid is then sprayed via an atomizer into a cool air stream, whereupon the droplets solidify and form particles. The system is therefore similar to conventional spray drying except that no solvent is involved and solidification occurs via cooling rather than heating and evaporation. The principles of the method are illustrated in Figure 1 a and b, with the latter showing the lab-scale system used in the studies described here. Scale-up to industrial utility is achievable and has been demonstrated in the manufacture of some marketed products.
The basic principles of spray chilling are, as stated above extremely simple, although inevitably there are numerous opportunities and subtleties associated with optimizing the process; the interested reader is referred to a number of sources, which discuss the processing aspects in more detail [11-19]. In brief, the technique involves the mixing of drug and lipid in a heated pressurized container such that the system is brought into the molten state. The resulting liquid is then sprayed via an atomizer into a cool air stream, whereupon the droplets solidify and form particles. The system is therefore similar to conventional spray drying except that no solvent is involved and solidification occurs via cooling rather than heating and evaporation. The principles of the method are illustrated in Figure 1 a and b, with the latter showing the lab-scale system used in the studies described here. Scale-up to industrial utility is achievable and has been demonstrated in the manufacture of some marketed products.
Inevitably, there are issues that need to be considered carefully in designing the process. The Passerini group have looked carefully at the atomization step in relation to particle structure [16,19], while we have developed an interest in attempting to predict which drug-lipid combinations the system is suited for, as our experience indicates that despite suitable melting point characteristics the technique is not universally applicable, with many systems simply failing to form discreet particles. We have developed a thermorheological test whereby we monitor the viscoelastic properties (or simply the viscosity, which is adequate for this purpose) as the system cools from the melt [20]. If the viscosity increases dramatically on cooling to the region associated with the melting of the lipid then the implication is that the drug is not influencing the crystallization behavior of the lipid and the system should cool to the solid state during the manufacturing process. 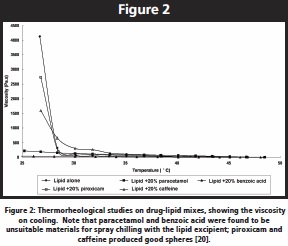 If, however, the system demonstrates extensive cooling to below the melting point of the lipid then the drug is clearly inhibiting solidification of the lipid. This is demonstrated in Figure 2 for a range of drugs in a solid lipid excipient, which mainly contains glycerides and fatty acid esters of polyethylene glycol; a direct correlation was noted between the tendency for the viscosity to increase and the ability of the mixture to form particles on spray chilling [20]. While a simple predictive test, this also illustrates the extremely important point that applies to all drug dispersion systems, namely that it is essential to understand the physical relationship between the drug and carrier if one is to manufacture dosage forms in a predictable and rational manner.
If, however, the system demonstrates extensive cooling to below the melting point of the lipid then the drug is clearly inhibiting solidification of the lipid. This is demonstrated in Figure 2 for a range of drugs in a solid lipid excipient, which mainly contains glycerides and fatty acid esters of polyethylene glycol; a direct correlation was noted between the tendency for the viscosity to increase and the ability of the mixture to form particles on spray chilling [20]. While a simple predictive test, this also illustrates the extremely important point that applies to all drug dispersion systems, namely that it is essential to understand the physical relationship between the drug and carrier if one is to manufacture dosage forms in a predictable and rational manner.
Taste Masking Microspheres
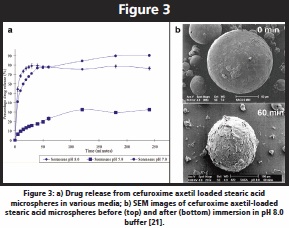
The thrust of the initial work within our group using this technique was concerned with the development of taste masking microspheres. In brief, our interest lay in understanding why spray chilled stearic acid microspheres containing the drug cefuroxime axetil appeared to allow preparation of a suspension whereby the child patient was not able to taste the drug but the bioavailability was not adversely affected, indicating release in the lower gastrointestinal tract. Our initial studies focused on the characterization of the spheres before and after immersion in a range of simulated gastric fluids. Of particular note was the observation that immersion in higher pH media resulted not only in faster release (Figure 3a) but also what appeared to be compromise of the surface integrity of the particles (Figure 3b) [21-23]. Our subsequent studies explored the effect of lipid composition on the structure and release, bearing in mind that commercial stearic acid is in fact largely a mix of stearic (C18) and palmitic acids (C16), with a range of lower chain length lipids present as minor components [24-26].
 In many respects the study evolved into a pertinent illustration of the interrelationship between pharmaceutical technology and materials science, as it rapidly became apparent that simple explanations for the release behavior such as ionization of the stearic acid in alkali conditions were not correct. In fact, there were clearly twin complexities in terms of the structure of the spheres themselves and the nature of their interaction with the media. In terms of structure, we believe that the stearic and palmitic acids form a eutectic mixture on spray chilling but, crucially, on immersion in alkali media a further structure is formed, as evidenced by the appearance of new peaks in the differential scanning calorimetry (DSC) data which do not correspond to simple salt formation. We ascribed this to the formation of acid soaps, these being complexes between the free acid and salt forms of the lipid [24-26] which form at pH values intermediate between those favoring negligible or full ionization. Such systems are well known in the detergent field but to our knowledge are not widely recognized within pharmaceutical formulation. To add further to the complexity, we also performed monolayer studies to attempt to understand the nature of the lipidmedia interaction at a molecular level. We used simple compression studies but also utilized technique such as Brewster Angle Microscopy, a laser imaging technique, which allows identification of discreet compositional regions on monolayers as well as neutron scattering whereby we are able to trace the presence and dimensions of labeled lipid molecules as a function of time and compression in complex mixes [25]. The overall conclusion from these extensive studies was that the lipids form eutectic structures on initial formation, which determine the ease with which the lipid could interaction with the media. On immersion in the alkali media, two related processes appear to be at play. Firstly, the lipids form acid soaps, which disrupt the integrity of the spheres and secondly the lower chain length lipids leach into the media, thereby allowing drug release. If nothing else these studies illustrate how a seemingly simple two component system can behave in a manner which is highly complex, yet understanding this complexity is essential in order to identify and control the associated release mechanisms.
In many respects the study evolved into a pertinent illustration of the interrelationship between pharmaceutical technology and materials science, as it rapidly became apparent that simple explanations for the release behavior such as ionization of the stearic acid in alkali conditions were not correct. In fact, there were clearly twin complexities in terms of the structure of the spheres themselves and the nature of their interaction with the media. In terms of structure, we believe that the stearic and palmitic acids form a eutectic mixture on spray chilling but, crucially, on immersion in alkali media a further structure is formed, as evidenced by the appearance of new peaks in the differential scanning calorimetry (DSC) data which do not correspond to simple salt formation. We ascribed this to the formation of acid soaps, these being complexes between the free acid and salt forms of the lipid [24-26] which form at pH values intermediate between those favoring negligible or full ionization. Such systems are well known in the detergent field but to our knowledge are not widely recognized within pharmaceutical formulation. To add further to the complexity, we also performed monolayer studies to attempt to understand the nature of the lipidmedia interaction at a molecular level. We used simple compression studies but also utilized technique such as Brewster Angle Microscopy, a laser imaging technique, which allows identification of discreet compositional regions on monolayers as well as neutron scattering whereby we are able to trace the presence and dimensions of labeled lipid molecules as a function of time and compression in complex mixes [25]. The overall conclusion from these extensive studies was that the lipids form eutectic structures on initial formation, which determine the ease with which the lipid could interaction with the media. On immersion in the alkali media, two related processes appear to be at play. Firstly, the lipids form acid soaps, which disrupt the integrity of the spheres and secondly the lower chain length lipids leach into the media, thereby allowing drug release. If nothing else these studies illustrate how a seemingly simple two component system can behave in a manner which is highly complex, yet understanding this complexity is essential in order to identify and control the associated release mechanisms.
Fast-release Lipid Particles
While solid lipid systems are often associated with slow release, there are many circumstances whereby they are able to elicit fast release for poorly bioavailable drugs. A well known example of this is the class of new self-emulsifying solid/semi-solid lipid excipients, which forms an emulsion on contact with water which may in turn lead to improved dissolution and bioavailability [27]. What is less widely appreciated is that lipids which are in themselves not associated with fast release may also improve dissolution, an example being a lipid excipient (mainly containing glycerides of hydrogenated palm oil and mono and diesters of PEG 1500) which is typically associated with sustained release.  However, it has been noted [28] that preparation in particulate form may have precisely the opposite outcome, hence opening up the possibility of using this material for poorly water-soluble drugs. In our own study [20] we examined the incorporation of piroxicam in the lipid excipient spray chilled particles, not just in terms of the release profile but also from the viewpoint of trying to identify the release mechanism. Returning to an earlier theme, one issue we were keen to explore was whether the drug was ‘dissolved’ in the lipid in the solid state or whether it was dispersed in particulate form; Figure 4 illustrates the use of hot stage microscopy in this regard whereby by rapidly heating the spheres we are able to identify the presence of drug particles in the molten system. It should be noted that caution is required when using DSC for the same objective, as dissolution of the particulate drug in the molten lipid on heating can be difficult to detect and hence may lead to the erroneous conclusion that the drug was dissolved in the lipid initially. Note that this dissolution effect can actually be seen in Figure 4; if one observes the 60oC image it is apparent that less drug appears to be present than at lower temperatures, despite our being well below the melting point of the drug.
However, it has been noted [28] that preparation in particulate form may have precisely the opposite outcome, hence opening up the possibility of using this material for poorly water-soluble drugs. In our own study [20] we examined the incorporation of piroxicam in the lipid excipient spray chilled particles, not just in terms of the release profile but also from the viewpoint of trying to identify the release mechanism. Returning to an earlier theme, one issue we were keen to explore was whether the drug was ‘dissolved’ in the lipid in the solid state or whether it was dispersed in particulate form; Figure 4 illustrates the use of hot stage microscopy in this regard whereby by rapidly heating the spheres we are able to identify the presence of drug particles in the molten system. It should be noted that caution is required when using DSC for the same objective, as dissolution of the particulate drug in the molten lipid on heating can be difficult to detect and hence may lead to the erroneous conclusion that the drug was dissolved in the lipid initially. Note that this dissolution effect can actually be seen in Figure 4; if one observes the 60oC image it is apparent that less drug appears to be present than at lower temperatures, despite our being well below the melting point of the drug.
A particularly useful technique for the study of the release mechanism turned out to be simple optical microscopy, whereby we were able to demonstrate that on immersion in water the drug particles appeared to remain remarkably stationary for extended periods of time. This led us to the hypothesis that the lipid may be forming some type of network in water in which the drug particles were trapped but from which dissolution was enhanced due to lack of aggregation. Small angle X-ray diffraction studies confirmed the generation of a cubic phase system by the hydrated lipid excipient, this being a high viscosity but also high water permeability structure which would allow both drug entrapment and water access [20]. Our hypothesis is therefore that the lipid is holding the drug particles separately in a viscous network, through which water nevertheless moves freely thus enabling dissolution to occur.
Conclusions
Spray chilling of lipids represents an interesting and potentially highly useful means of preparing solid lipid dosage forms, having the advantages of simplicity, speed, reasonably low cost once the equipment is established and absence of solvents. However, once again the principle that even two-component systems can be highly complex is manifest here, both in terms of understanding the processing parameters in relation to subsequent structure and also the performance and behavior of that product. Nevertheless, given the high proportion of ‘difficult’ drugs that are now being generated it is essential to look to new strategies in formulation science to get such drugs on the market, hence it is very likely that there is a potentially important place for this technology amongst the options now available to the industry.
Acknowledgements
There were numerous people involved in the establishment of the spray chilling initiative within our group. Hazel Robson initiated the work on both spray chilling and taste masking during her PhD and Dr. Simon Gaisford built and commissioned the equipment as well as performing the early formulation studies on which subsequent work was based prior to Sheng Qi joining the group; she has since led the work in the field within the group. Throughout this period, we wish to thank David Deutsch and GlaxoSmithKline for their continued support and advice.
Reference
1. Charman WN. Lipids, lipophilic drugs, and oral drug delivery – some emerging concepts. J. Pharm. Sci. 2000, 89:967-978.
2. Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: Optimising the oral delivery of lipophilic drugs. Nature Reviews Drug Discovery. 2007, 6:231-248.
3. Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv. Rev. 2007, 59(7): 667-676.
4. Pouton CW, Porter CJH. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv. Drug. Deliv. Rev. 2008, 60:625-637.
5. Porter CJH, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv. Rev. 2008, 60:673-691.
6. Wang PY. Palmitic acid as an excipient in implants for sustained release of insulin. Biomaterials 1991, 12(1): 57-62.
7. Hassan EE, Eshra AG, Nada AH. Formulation of prolonged release lipid micropellets by emulsion congealing: Optimization of ketoprofen entrapment and release. Int. J. Pharm. 1995, 121(2):149-155.
8. Yamaguchi T. Lipid microspheres as drug carriers: a pharmaceutical point of view. Adv. Drug. Deliv. Rev. 1996, 20(2-3):117-130.
9. Grassi M, Voinovich D, Franceschinis E, Perissutti B, Filipovic-Grcic J. Theoretical and experimental study on theophylline release from stearic acid cylindrical delivery systems. J. Control. Release. 2003, 92(3): 275-289.
10. Hauss DJ. (ed.) Oral lipid-formulations: enhancing the bioavailability of poorly water-soluble drugs. 2007, Informa HealthCare, New York.
11. Cusimano AG, Becker CH. Spray-congealed formulations of sulfaethylthiadiazole (SETD) and waxes for prolonged-release medication. J. Pharm. Sci. 1968, 57(7): 1104-1112.
12. John PM, Becker CH. Surfactant Effects on Spray-Congealed Formulations of Sulfaethyiazole-Wax. J. Pharm. Sci. 1968, 57: 584-589.
13. Yajima T, Nogata A, Demachi M, Umeki N, Itai S, Yunoki N, Nemoto M. Particle design for taste-masking using a spray congealing technique. Chem. Pharm. Bull. 1996, 44: 187-191.
14. Yajima T, Umeki N, Itai S. Optimum spray Congealing conditions for Masking the Bitter Taste of Clarithromycin in Wax Matrix. Chem. Pharm. Bull. 1999, 47: 220-225.
15. Yajima T, Umeki N, Itai S. Optimum spray congealing conditions for masking the bitter taste of clarithomycin in wax matrix. Chem. Pharm. Bull. 1999, 47: 220–225
16. Rodriguez L, Passerini N, Cavallari C, Cini M, Sancin P, Fini A. Description and preliminary evaluation of a new ultrasonic atomizer for spray congealing processes. Int. J. Pharm. 1999, 183:133–143.
17. Maschke A, Becker C, Eyrich D, Kiermaier J, Blunk T, Gopferich A. Development of a spray congealing process for the preparation of insulin loaded lipid microparticles and characterization thereof. Eur. J. Pharm. Biopharm. 2007, 65:175–187.
18. Hincal AA, Kaş HS. Preparation of micropelles by spray congealing in Multiparticulate oral drug delivery, Isaac Ghebre-Sellassie (Ed.), Marcel Dekker, New York, 1994, page 17-34.
19. Passerini N, Qi S, Albertini B, Grassi M, Rodriguez L, Craig DQM. Solid lipid microparticles produced by spray congealing: Influence of the atomizer on microparticle characteristics and mathematical modeling of the drug release. J. Pharm. Sci. 2010, 99(2): 916-931.
20. Qi S, Marchaud D, Craig DQM. An investigation into the mechanism of dissolution rate enhancement of poorly water-soluble drugs from spray chilled gelucire 50/13 microspheres. J. Pharm. Sci. 2010, 99(1): 262-274.
21. Robson HJ, Craig DQM, Deutsch D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres: Part 1: The influence of the dissolution medium on the drug release profile and the physical integrity of the microspheres. Int. J. Pharm. 1999, 190(2): 183-192.
22. Robson HJ, Craig DQM, Deutsch D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres. II. The effects of buffer composition on drug release. Int. J. Pharm. 2000, 195(1-2): 137-145.
23. Robson HJ, Craig DQM, Deutsch D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres. III. The use of DSC and HSDSC as means of characterising the interaction of the microspheres with buffered media. Int. J. Pharm. 2000, 201(2): 211-219.
24. Qi S, Deutsch D, Craig DQM. An investigation into the interaction between taste masking fatty acid microspheres and alkaline buffer using thermal and spectroscopic analysis. J. Pharm. Sci. 2006, 95(5): 1022-1028.
25. Qi S, Roser SJ, Deutsch D, Barker SA, Craig DQM. A laser imaging and neutron reflection investigation into the monolayer behaviour of fatty acids used for taste masking microspheres. J. Pharm. Sci. 2008, 97(5): 1864-1877.
26. Qi S, Deutsch D, Craig DQM. An investigation into the mechanisms of drug release from taste-masking fatty acid microspheres. J. Pharm. Sci. 2008, 97(9): 3842-3854.
27. Barker SA, Yap SP, Yuen KH, McCoy CP, Murphy JR, Craig DQM. An investigation into the structure and bioavailability of alpha-tocopherol dispersions in Gelucire 44/14. J. Control. Release. 2003, 91 (3): 477-488.
28. Passerini N, Perissutti B, Moneghini M, Voinovich D, Albertini B, Cavallari C, Rodriguez L. Characterization of carbamazepine-Gelucire 50/13 microparticles prepared by a spray-congealing process using ultrasounds. J. Pharm. Sci. 2002, 91(3):699-707.
Duncan Craig holds the Chair in Pharmaceutics and is Head of the School of Pharmacy, University of East Anglia. His research involves the physical characterisation of dosage forms in relation to performance using a range of thermal, rheological and imaging approaches.
Sheng Qi is a lecturer in Pharmaceutics at the School of Pharmacy, University of East Anglia. Her research mainly involves the development of lipid based dosage forms and the use of novel physicochemical characterisation approaches for solid dosage forms.