Abstract
Standard dissolution testing employs sink conditions and thus is not predictive for absorbability of poorly soluble drugs in vivo. Therefore special dissolution tests for optimization of formulations were developed: most suitable is to dissolve the highest assumed dose in man in a volume of 250 ml at the pH of minimal solubility in the range of pH 1 – 6 using low buffer strengths. Furthermore a test system for extended release of poorly soluble drugs using octanol as absorption phase was developed.
Introduction
Standard dissolution testing employs well defined USP methods, volumes of 500 or 900 ml and sink conditions, that means final dissolution reaches 100%. To obtain sink conditions, weak bases are dissolved mostly at acidic pH, weak acids at slightly alkaline pH. These tests are a suitable tool for quality control, but absorption of poorly soluble drugs can hardly be predicted by standard dissolution testing, as neither volumes nor the pH conditions reflect practical intake of drugs in man. As percentage of BCS class II & IV drugs (Active Pharmaceutical Ingredients, API’s) increased to about 90% (Ref 1) over the last years, special dissolution methods predicting absorbability of difficult APIs in man have to be employed.
General Aspects on Formulation and Dissolution Strategy for Poorly Soluble Drugs
Adequate formulation of difficult APIs requires a clear understanding of therapeutic needs, physiology of the GI tract, pharmacokinetics & physicochemical properties of the API and high throughput in formulation and dissolution work. Thus dissolution is an integral part of development, no “self-purpose”; dissolution testing strategy has to be adapted to the special needs of the API. From the therapeutic needs the most suitable pharmacokinetic profile with regard to e.g. rapid onset of action, maintenance of therapeutically active plasma concentrations etc. can be defined. From these data and the physicochemical properties a suitable dissolution test system for optimization of formulations can be defined. Dissolution is prerequisite for absorption after oral intake. For poorly soluble drugs dissolution is highly improbable with conventional formulations. Goal of formulation for these BCS II and IV APIs is to make them behave as BCS I & III compounds, by applying special excipients and/or formulation technologies in order to improve dissolution. To achieve this in the early phase of API development, formulation as well as testing strategies have to be performed with high throughput and minimal amounts of API.
High Throughput Dissolution Testing Predicting Absorbability In Man
The following dissolution test system achieved adequate predictability:
- The concentration of the API is equivalent to highest assumed dose in man in a volume of 250 ml. This reflects BCS considerations and also intake of drugs in phase I studies.
- The pH of the buffer systems is adjusted to the pH of lowest solubility of the API in the range of pH 1 to 6 (if feasible).
- The buffer strength is 0.1 M or less, that means no “robust” buffers are used.
- The volume of dissolution medium: 0.2 to 200 ml depending on dissolution method in order to safe amounts of API.
- Detection mode is UV-measurement with turbidity correction.
Necessity and Mode of Turbidity Correction
 With high dose numbers of the API, no complete dissolution is obtained in most cases. Therefore UV signal is an overlay of “true” UV spectrum and turbidity.
With high dose numbers of the API, no complete dissolution is obtained in most cases. Therefore UV signal is an overlay of “true” UV spectrum and turbidity.
Turbidity could be removed by filtration or centrifugation, but in some cases the supersaturation is destroyed by these procedures, resulting in false negative results.
As prediction of absorption in man represents primary goal of the dissolution, not “correct” dissolution data, UV-absorbance after correction of turbidity is used.
The underlying working hypothesisis is: “the UV-absorbance part which reflects the correct UV-spectrum of the API is the absorbable proportion of total amount of API.”
The correction for turbidity can be done as follows:
- Subtract absorption at a higher wavelength, where dissolved API has no UV-absorbance (this is correct only when absorbance of turbidity is independent from wavelength)
- extrapolate absorption at API maximum from shape of turbidity at higher wavelength, where dissolved API has no UV-absorbance (use software options like scatter correction or Excel)
- Use derivate methods implemented in dissolution software
- Chose linear, squared or exponential function to calculate spectrum of turbidity by absorption measurement at different wavelength and calculate “true” UV-absorbance, e.g. by using the function “solver” of Excel.
Considerations for Weak Bases
As solubility is frequently adequate at acidic pH, absorbability of conventional formulations is frequently given under acidic stomach conditions.
As non-acidic stomach conditions however occur frequently in elderly or with antacid comedications, achievement of pH independent dissolution is to be preferred in many indications, especially when elderly patients are the target population.
Considerations for Weak Acids
As solubility is frequently adequate only at slightly alkaline pH, dissolution and absorbability of conventional formulations is given even in a best case situation only in the lower parts of the GI tract. Therefore they show an extended release absorption profile even if dissolution of the formulation is rapid at alkaline pH.
If a rapid onset of action is mandatory, achievement of pH independent dissolution is necessary in all cases.
Conduct of Formulation Screening - Experimental Setup
Most poorly soluble drugs show pH dependent solubility. Best dissolution for these APIs is usually obtained using combinations of pH modifiers, solubilizers and supersaturating excipients. In order to achieve high throughput, stock solutions are prepared for API and all excipients in water, the “formulations” are prepared by mixing API and excipients directly in the dissolution vessels and subsequent evaporation. A schematic presentation of mixtures is given in Table 1. 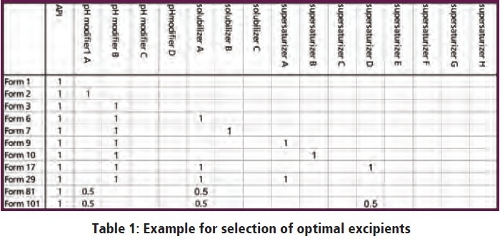
Dissolution is observed regarding dissolution speed, degree and time to reprecipitation in buffers of minimal solubility of the API at 37 °C. It is evident that the API shows poor dissolution, a suitable pH modifier achieves good initial dissolution but also marked reprecipitation which however can be prevented in the combination with a suitable supersaturizing excipient.
Figure 2 shows some dissolution results from a formulation screening of a poorly soluble week base at pH 6.
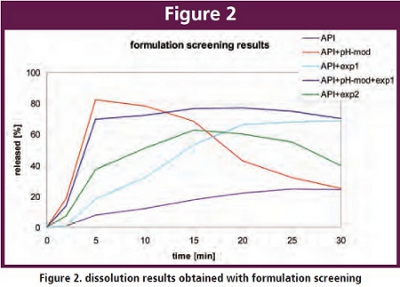 It is evident that the API shows poor dissolution, a suitable pH modifier achieves good initial dissolution but also marked reprecipitation which however can be prevented in the combination with a suitable supersaturizing excipient.
It is evident that the API shows poor dissolution, a suitable pH modifier achieves good initial dissolution but also marked reprecipitation which however can be prevented in the combination with a suitable supersaturizing excipient.
Development of Solid Formulations
The outcome of the previous steps is an optimized formulation with regard to achieve dissolution and supersaturation. Process development and other necessary functions e.g. rapid disintegration however are still completely missing. Therefore a solid dosage form suitable for oral intake in patients has to be developed. As this also may require a large number of experiments, the first manufacturing steps should also be performed at small scale. 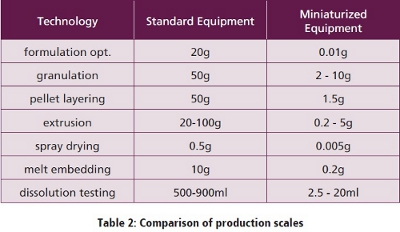 A few years ago no small scale equipment for production of formulations or dissolution testing was available, small scale equipment was developed by R. Brickl at Boehringer Ingelheim. A survey on the downsizing achieved is given in Table 2. In the meantime also several other companies developed small scale equipment.
A few years ago no small scale equipment for production of formulations or dissolution testing was available, small scale equipment was developed by R. Brickl at Boehringer Ingelheim. A survey on the downsizing achieved is given in Table 2. In the meantime also several other companies developed small scale equipment.
Miniaturized Dissolution Tester
A small scale equipment with working volumes of 10 – 20 ml was developed which can use paddles or baskets and thus is very similar to conventional USP methods was developed. By a slight adjustment of rotation speed, nearly identical dissolution results compared to a conventional USP I method of batches with different release characteristics of a week base at ph 6.0 (at this pH solubility of the API is close to zero) could be achieved. 
Dissolution Model for Extended Release Formulations
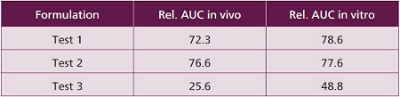
When an extended release formulation was developed of a weak base soluble only at acidic pH, no correlation of in vivo absorption to a variety of conventional in vitro dissolution tests could be found. 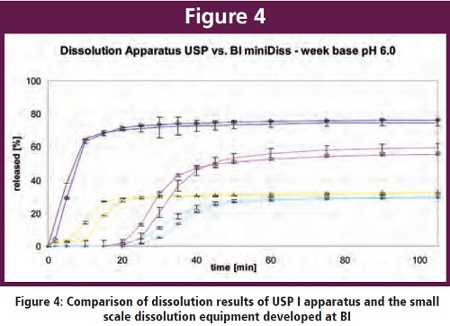 This was only feasible when octanol was added to the buffer system as an absorption phase. The best correlation was obtained when 550m ml of phosphate buffer and 100 ml octanol were used as medium, 50 rpm and paddle were employed. PH 2 was used from 0-1 hrs, constant pH was 5.5 from 1-3 hrs (achieved by autotitration) flexible pH was 5.5 from 3-5 hrs and 6.8 from 5 – 24 hrs. A comparison of in vitro and in vivo data is given in Table 3. It obvious that in vitro performance is quite predictive for in vivo behavior.
This was only feasible when octanol was added to the buffer system as an absorption phase. The best correlation was obtained when 550m ml of phosphate buffer and 100 ml octanol were used as medium, 50 rpm and paddle were employed. PH 2 was used from 0-1 hrs, constant pH was 5.5 from 1-3 hrs (achieved by autotitration) flexible pH was 5.5 from 3-5 hrs and 6.8 from 5 – 24 hrs. A comparison of in vitro and in vivo data is given in Table 3. It obvious that in vitro performance is quite predictive for in vivo behavior.
References
- Ref 1 David Hauss, Oral Lipid Based Drug Delivery, 2007
Rolf Brickl, PhD was born October 8, 1944 in Munich, Germany. He received his Diploma in 1969 and his Ph.D. in 1971 in Physical Chemistry at Tachnical University Munich.
He has worked at Boehringer Ingelheim Pharma GMBH & Co. KG since 1972 in the fields of Dermatological Research, Human Pharmacokinetics and Pharmaceutical Research and Development. Rolf is a scientific expert in formulation and kinetics of extended release formulations and poorly soluble drugs, development of high throughput formulation technologies and dissolution technologies, development of miniaturized formulation, and dissolution equipment.
He has created many patents, written papers, and given presentations at international meetings, and he is the winner of Boehringer Ingelheim Research and Development Award in 2004 and of German Development Award Boehringer Ingelheim in 2008.