Introduction
Cleaning validation is a GMP requirement [1-2] in the pharmaceutical manufacturing of drug substances and drug products. Cleaning verification (CV) must be performed to demonstrate the “cleanliness” of the production equipment [3-4] at the completion of each manufacturing step by visual inspection and the analysis of cleaning swabs or rinse solutions to confirm that the active pharmaceutical ingredient (API) has been adequately removed to pre-established acceptance limits [4-5]. Cleaning validation is a multifunctional program in late-stage drug development and commercial production and requires a thorough understanding of equipment design, contact surfaces, product solubility, and other associated properties in order to establish practical cleaning protocols, acceptance limits, and analytical procedures [4, 6]. The analytical procedures typically used have been reviewed by Pack [4]. They can be non-specific (e.g., gravimetric (residual on evaporation or ROE)) or total organic carbon (TOC)) or product-specific techniques such as ultraviolet spectrometry (UV), ion mobility spectrometry (IMS), or high-performance liquid chromatography (HPLC) using UV, evaporative light scattering (ELSD), charged aerosol detection (CAD), mass spectrometry (MS), or other detectors [4, 6-8].
In this paper, we describe the development and qualification of a 10-min generic HPLC/UV platform method for cleaning verification of diversified API rinse solutions at concentration of 0.2 to 10 μg/ mL. Sample preparation is a simple dilution with water to allow an injection volume of 20 μL. For highly potent compounds requiring a lower limit of quantitation (LOQ), we demonstrate several approaches to enhance sensitivity to low ng/mL by large volume injections, sample concentrations, and the use of newly developed long-path UV flowcells. Using UHPLC equipment with low dwell volumes, similar performance to the 10-min generic method can be achieved in 1.5-min by sub-2-μm or sub-3-μmcore-shell columns [9, 10]. Finally, we illustrate the application of chromatographic principles in selecting columns and operating parameters to allow rapid development of a practical method with requisite performance (sensitivity, peak capacity (Pc) and analysis time (t)) for intended use [10, 11]. This generic platform technology is readily adaptable to analysis of swabbing/rinse solutions of many pharmaceuticals. Potential benefits of having a single generic method include quicker implementation, reduced validation efforts, and operation under open-access environment with lessexperienced staff.
Experimental
HPLC systems with pressure limits of 400 bar (6,000 psi) equipped with either binary or quaternary pumps, autosamplers, column ovens and photo diode array (PDA) detectors with 10-mm flowcells were used. An ultra-high pressure LC (UHPLC) system with a pressure limit of 1200 bar (18,000 psi) with a binary pump, an autosampler, a column oven and a PDA detector with a standard 10-mm or an extended pathlength 60-mm flowcell was also used. The dwell volume of the UHPLC system was measured to be 240 μL (with a 100-μL mixer) vs. ~1000 μL for the standard HPLC systems. The mass spectrometer used was either a single quadrupole MS (SQ-MS) using single ion monitoring (SIM) with electrospray ionization (ESI) or a triple quadrupole MS (TQ-MS) using multiple reaction monitoring (MRM) with ESI. Mobile phase A (MPA) was 20 mM ammonium acetate at pH 3.7 in water for the initial method or 0.05% trifluoroacetic acid (TFA) in water for the final standard methods. Mobile phase B (MPB) was 0.05% formic acid (FA) in acetonitrile (ACN) for the initial method or 0.05% TFA in ACN for the final methods. The columns used were a C18 (50 mm x 3.0 mm id, 3.5 μm) for the initial and standard methods, a C18 (50 mm x 2.1 mm id, 1.7 μm) for the UHPLC method and a C18 (30 mm x 2.1 mm id, 2.7 μm core-shell) for the Fast LC method [9].

Initial Method Development Strategies and Rationales
We initiated our cleaning verification method development to support a pending internal 10-kg API synthesis campaign in our process lab. We estimated an initial acceptance limit of 0.5 to 1 μg/ mL in the rinse solution for this compound based on “the 10 ppm method” [4]. The calculation assumes a worst-case scenario of a next-batch size of 1 kg which yields a total acceptable residue value of 10 mg in the 1-kg batch in a 100-L vessel. Using total volume of rinse solvent of 1/10 of the vessel size, a rinse solution level of 10 mg in 10 L or 1 μg/mL is obtained (or a 0.5 μg/mL level allowing for a 1:1 dilution ). Methanol (MeOH) was chosen to be the rinse solvent due to its high solubility for this compound (coded GNE A).
The initial starting point for method development stemmed from the existing stability-indicating assay/impurity method which uses a 150 mm x 4.6 mm, 3 μm C18 column with a 20 mM ammonium acetate/acetonitrile solvent program. This multi-segment assay gradient method separates all impurities and degradants and has a run time of 42 min and a limit of quantitation (LOQ) of 2.5 μg/mL. To improve sensitivity and analysis time, we adopted the same mobile phases with a shorter and narrower column (50 mm x 3.0 mm, 3.5 μm). The gradient method was selected over the more traditional isocratic methods for cleaning verification (CV) applications for several reasons: 1. A single generic CV method amendable to multiple APIs is desired. 2. Gradient methods have better sensitivity (sharper peaks and higher injection volumes) [10]. 3. Gradient methods have higher Pc than isocratic methods [10, 11]. We maintained the same flow rate of 1 mL/min and detection wavelength of 280 nm (λmax of GNE A). We selected a gradient range of 5% to 70% MPB, based on one of our generic in-process control (IPC) method from our process chemistry lab which was applicable to many new chemical entities (NCEs) [12].
The choice of a smaller diameter column at 3.0 mm id vs. a more conventional id of 4.6 mm (2.2X reduction of column void volume (Vm), allows the attainment of high mass sensitivity or lower injection volumes by the same factor [10]. The use of 1 mL/min (equivalent to a flow of 2.2 mL/min on the 4.6-mm id column) results in higher Pc for better method specificities [10, 11]. The method development process is a balancing act of numerous operating parameters selected in this case to maximize detection sensitivity while maintaining reasonable Pc and analysis time (t) which is typically controlled by gradient time (tG).
Table 1 (column one) lists the initial HPLC conditions and pertinent performance parameters. Utilizing HPLC fundamentals plus a few hours of laboratory time, we were able to achieve an LOQ of <0.1 μg/ mL with an injection volume of 20 μL and a run time 6 min. Figure 1 (left side), shows chromatograms of the blank and methanol solutions of GNE A at concentrations of 0.1, 1.0 and 10 μg/mL. Considerable peak fronting was observed from the direct injections of strong solvent (20 μL of MeOH). By diluting of the MeOH solutions 1:1 with MPA, excellent peak shapes and improved detection sensitivity were observed in Figure 1 (right-side chromatograms). The MS-compatible mobile phases allowed us to use the same method for MS detection. As expected, considerable lower LOQs of 3 ng/mL (SQ-MS, SIM) and 0.02 ng/mL (TQ-MS, MRM) respectively were obtained. Nevertheless, UV detection is preferred for CV analyses due to its wide availability in the QC labs, ease of use, robustness, and adequate sensitivity/ specificity for this application.
Approaches to Lower Limits of Quantitation (LoQs) for Highly Potent Compounds
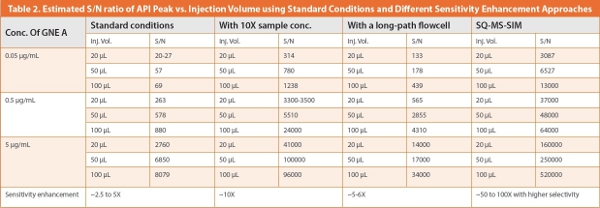
Next, we explored several approaches to enhance sensitivity for highly potent compounds. First, larger volumes of the diluted solutions were injected (20, 50 and 100 μL) yielding the expected increases of peak heights and signal/noise (S/N) ratio. Second, a 20-mL aliquot of the sample was evaporated to dryness and reconstituted with 2-mL of aliquot of 20% MeOH in MPA to enrich API concentration by tenfold. Thirdly, we replaced the standard 10-mm UV flowcell with an extended pathlength 60-mm flowcell. Note that these extended-path UV flowcells utilizing the principle of total internal reflectance can increase sensitivity significantly with some band-broadening and are available from many UHPLC manufacturers. Reported values for the standard deviation of dispersion (σv) the two flowcells are 1.0 and 4 μL respectively. Results of these approaches are summarized in Table 2 showing the generally expected increase of S/N ratio. These approaches can be used singly or in combination to increase sensitivity by 5 to 250 fold to support CV applications of highly potent compounds using the standard HPLC/UV method. Note that sample concentration (to dryness) is required if a chromophoric rinse solution such as acetone is used. In all cases, the signals from the API and any interfering compounds in the rinse samples are equally increased by all of these approaches. Therefore, they would not work if interfering peaks in the sample (e.g., interferences of impurities from the vessels, swabs or rinse solvents) were the limiting factors.

Method Revisions to Accommodate Multiple APIs and Method Qualification
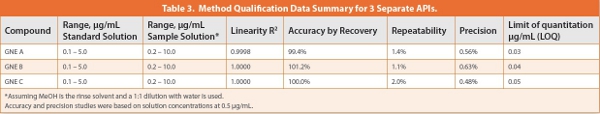
We further fine-tuned method conditions into a more general platform methodology for multiple APIs. To support two additional upcoming campaigns in the process labs (GNE B and C), we prepared a set of API solutions for all three NCEs at concentrations of 0.1 to 5 μg/mL in a 50% MeOH/water diluent. This diluent represents a reasonable compromise of solubilizing power for diversified compounds and chromatographic solvent strength to minimize peak shape problems during analysis. We kept the same column but made changes to the mobile phases and gradient conditions shown in Table 1 (column 2). A simpler MPA of 0.05% TFA was used which offers slightly higher retention for basic compounds (due to ion-paring effect) [10]. The UV detection wavelength was set to 265 nm, a compromise wavelength selected to yield roughly equal sensitivities for the three compounds (GNE A , B and C) with λmax of 280, 300 and 250 nm respectively. Figure 2 shows chromatograms of the blank and standard solutions at nominal concentrations of 0.1, 0.2, 0.5 and 1.0 μg/mL. Method qualification data to confirm the suitability of the method for intended use are summarized in Table 3.
Figure 3 shows overlaid chromatograms of seven additional NCEs in our clinical developmental portfolio at 0.1 μg/mL using the standard method. Note that different monitoring wavelengths were used for these diversified molecules. Nevertheless, the generic methodology appears to work well for all ten NCEs with adequate sensitivity. Peak shape issues (fronting) were observed with two early eluting compounds (GNE H and I) which may be remedied by using smaller injection volumes or weaker diluents.
Adaptation to Faster Analysis Using sub-2 μm and sub-3-μm Core-shell Columns
Finally, we explored the use of modern HPLC columns under highthroughput screening (HTS) conditions to reduce the analysis time to ~1.5 min [10]. The HPLC conditions and performance parameters are shown in Table 1 (columns 3 and 4). The sub-2-μm column has a pressure drop of 750 bar or 11,000 psi at 1.5 mL/min and requires the use of UHPLC systems, while the shorter 30-mm long, 2.7-μm core shell column has similar performance operating at 310 bar or 4,500 psi (chromatograms shown in Figure 4). This Fast LC method using core-shell columns is recommended for CV applications requiring rapid turnarounds since the Pc is comparable to the UHPLC method. It operates at much lower back pressures and is therefore compatible to conventional HPLC systems from the pressure limit standpoint, though analysis times (t) and peak widths would be higher due to higher dwell volumes and system dispersions [10].

Results and Discussions
 The HPLC method development process is complex due to the large number of controlling factors (column, mobile phase, operating, instrumental) affecting retention, resolution, selectivity, sensitivity, speed, peak shape, and other method performance parameters [10-11]. This case study illustrates the application of modern chromatographic principles in the rapid development of a single generic CV method for sensitive quantitation of multiple drug candidates. The key performance parameters are sensitivity, accuracy, and adaptability to multiple APIs. Here, we selected a C18 column with a conventional acidic mobile phase with a broad acetonitrile gradient [10]. The standard method uses a short, mini-bore column (50 mm x 3.0mm, 3.5 μm) at 1 mL/min to achieve a run time of 7 min with an injection volume of 20 μL in a 1:1 MeOH/water diluent. Up to 100 μL of injection volume appears to be well accommodated for most NCEs and can be used with approaches such as sample enrichment or the use of a long-path flow cell to extend LOQs to low ng/mL levels. Other more universal detection approaches such as low-UV detection (e.g., 200 nm with 0.05% phosphoric acid in water /ACN) or CAD detection for non-chromophoric compounds can also be considered [4]. Two faster methods using either 50 mm x 2.1 mm, 1.7 μm or 30 mm x 2.1 mm, 2.7 μm core-shell column at 1.5 mL/min @ 50°C with a run time (t) of 1.5 min were developed. These use high-throughput screening (HTS) conditions with ballistic 1-min gradients (tG = 1 min). The smaller columns and faster tG yield higher peak signals but also have pronounced baseline shifts and noise. Also, the smaller columns can accommodate lower injection volumes particularly when used with UHPLC systems with typical sample loop of 20 μL.
The HPLC method development process is complex due to the large number of controlling factors (column, mobile phase, operating, instrumental) affecting retention, resolution, selectivity, sensitivity, speed, peak shape, and other method performance parameters [10-11]. This case study illustrates the application of modern chromatographic principles in the rapid development of a single generic CV method for sensitive quantitation of multiple drug candidates. The key performance parameters are sensitivity, accuracy, and adaptability to multiple APIs. Here, we selected a C18 column with a conventional acidic mobile phase with a broad acetonitrile gradient [10]. The standard method uses a short, mini-bore column (50 mm x 3.0mm, 3.5 μm) at 1 mL/min to achieve a run time of 7 min with an injection volume of 20 μL in a 1:1 MeOH/water diluent. Up to 100 μL of injection volume appears to be well accommodated for most NCEs and can be used with approaches such as sample enrichment or the use of a long-path flow cell to extend LOQs to low ng/mL levels. Other more universal detection approaches such as low-UV detection (e.g., 200 nm with 0.05% phosphoric acid in water /ACN) or CAD detection for non-chromophoric compounds can also be considered [4]. Two faster methods using either 50 mm x 2.1 mm, 1.7 μm or 30 mm x 2.1 mm, 2.7 μm core-shell column at 1.5 mL/min @ 50°C with a run time (t) of 1.5 min were developed. These use high-throughput screening (HTS) conditions with ballistic 1-min gradients (tG = 1 min). The smaller columns and faster tG yield higher peak signals but also have pronounced baseline shifts and noise. Also, the smaller columns can accommodate lower injection volumes particularly when used with UHPLC systems with typical sample loop of 20 μL.
Summary and Conclusions
This paper describes the development of a single 10-min gradient HPLC/UV method for cleaning verification (CV) applications at levels of 0.2 to 10 μg/mL levels for many APIs. Additional sensitivity enhancements can be accomplished by large volume injections, sample enrichments and the use of long-path flowcells. Faster 1.5- min analyses can be performed with modern columns. We believe that a general adoption of generic platform technologies can greatly enhance analytical laboratory productivity by simplifying lab operation and reducing method development and validation efforts. This standard method is readily adaptable to swabbing or rinse solutions and possibly other applications such as IPC testing or non-stability-indicating potency assays of many pharmaceuticals.
Acknowledgments
The authors thank Nik Chetwyn, Mohammad Al-Sayah, Chris Goretski, Jessica Defreese, Sigrid Hubbell, Chris Walsh, and David Stirling of Genentech, Davy Guillarme of U. Geneva, and Brian W. Pack of Eli Lilly for helpful discussions and suggestions.
References
1. Code of Federal Regulations, Title 21, Part 211 – Current Good Manufacturing Practice for Finished Pharmaceuticals – Section 67, Equipment cleaning and maintenance (21 CFR 211.67, U.S. Food and Drug Administration, Silver Spring, Maryland, revised April 2011).
2. ICH Harmonised Tripartite Guideline, Q7, Good Manufacturing Practice Guide for Active Pharmaceutical Ingredient, International Conference on Harmonisation, (ICH , Geneva, Switzerland November 2010).
3. Haider, S.I., Asif, E. S., Cleaning Validation Manual: A comprehensive guide for the pharmaceutical and biotechnology industries, (CRC Press, Taylor and Francis, Boca Raton, FL 2010).
4. Pack, B.W., Cleaning verification for highly potent compounds, in Formulation and analytical development for low-dose oral drug product, Zheng, J., ed., (John Wiley & Sons, Hoboken, NJ, 2009).
5. Sharnez. R., Strategies for developing a robust cleaning process: Part 1: application of quality by design to cleaning. Amer. Pharm. Review, 13 (5) (July/August, 2010).
6. Plasz, A. Cleaning validation using HPLC for analysis, in Handbook of pharmaceutical analysis by HPLC, Ahuja, S., Dong, M.W. eds., (Elsevier/Academic Press, Amsterdam, 2005, Chapter 16).
7. Galella, E., Jennings, S., Srikoti, M., Bonasso, E., Cleaning verification: method development and validation using ion mobility spectrometry, Pharm. Tech. 33(7), 60-63 (2009)
8. Liu, L., Pack, B.W., Cleaning verification assays for highly potent compounds by HPLC/MS: strategy, validation and long-term performance, J. Pharma. Biomed. Anal. 43, 1206-1212 (2007).
9. Ruta, J., Zurlino, D., Grivel, C., Heinisch, S., Veuthey, J.-L., Guillarme, D., Evaluation of columns packed with shell particles with compounds of pharmaceutical interest, J. Chromatogr. A, 1228, 221-231 (2012)
10. Dong, M.W., Modern HPLC for Practicing Scientists, Wiley-Interscience, Hoboken, NJ, 2006, Chapters 2, 3, 4 and 8.
11. Rasmussen, H.T., Li, W.Y, Redlich, D, Jimidar, M.I., Method development in early pharmaceutical development, In Handbook of Pharmaceutical Analysis by HPLC, Ahuja, S., Dong, M.W. eds., (Elsevier/Academic Press, Amsterdam, 2005, Chapter 6).
12. Garcia, R., Genentech, private communication, 2012.
Author Biographies
Michael W. Dong, Ph.D. is senior scientist in Small Molecule Analytical Chemistry and Quality Control (SMACQC), Small Molecule Drug Discovery, at Genentech in South San Francisco, where he is responsible for new technologies, automation and analytical support for multiple late-stage research projects. He holds a Ph.D. in Analytical Chemistry from City University of New York and a certificate in biotechnology from U. California at Santa Cruz. He has 80+ publications and 3 books including a bestseller in chromatography – Modern HPLC for practicing scientists, Wiley, 2006. He teaches short courses in HPLC/UHPLC, HPLC method development drug development process, and drug quality fundamentals for Amer. Chem. Society, Pittcon and Eastern Analytical Symposium.
Eileen X. Zhao is senior research associate in SMACQC in Genentech responsible for analytical development and QC of multiple early development projects. She holds a B.Sc. degree in Chemistry from Sichuan U. in China.
Derrick T. Yazzie is research associate in SMACQC in Genentech responsible for analytical development and QC of multiple early development projects. He holds a B.Sc. degree in Chemistry from Stanford U.
Christine C. Gu, Ph.D. is scientist in SMACQC in Genentech responsible for identification and structure elucidation of impurities and degradants in multiple development projects. She is a subject matter expert in mass spectrometry and holds a Ph.D. degree in Toxicology at U. California at Riverside.
Jackson D. Pellett, Ph.D. is scientist in SMACQC in Genentech responsible for leading analytical support for multiple early development projects, and for process chemistry and pharmaceutics labs. He is a subject matter expert in Quality by Design in drug development and holds a Ph.D. degree in analytical chemistry from U. Minnesota.