1 BASF Corporation, Tarrytown, NY (USA)
2 BASF SE, Ludwigshafen (Germany)
Introduction
Over 60% of new chemical entities that are poorly soluble qualify either as BCS Class II or Class IV and they provide challenges as well as opportunities to scientists working in formulation development [1]. The conventional solubilization approaches such as physical modifications of drug crystals (surface alteration of API, micronization or micro-milling) usually lead to a limited dissolution and solubility enhancement, but when developing a medium or high dosed formulation, the non-conventional formulation approaches are often required particularly when dealing with almost water-insoluble compounds usually characterized by a high melting point and/or very high lipophilicity [2].
Dosage requirements in the drug development also introduce opportunities to explore the other non-conventional formulation approaches for enhanced solubilization [3]. Liquid and solid dispersions, especially, are widely considered the alternative methods, which may require a range of factors for selecting one versus the other. Most importantly, the factors considered are: polymers, surfactants and solubilizers types, thermal stability, aqueous and organic solubility and compatibility, pH dependent invariability, and solubilization capability among others. In all, these formulation approaches are excipients-driven. With limited scope in the excipients’ selection, the industry is seeking new excipients to bring the drugs to the market faster and with a better performance. Furthermore, the development of safe and efficacious formulations (solid and liquid dispersions) requires complete evaluation of dosage stability, therapeutic efficacy and mitigation of food effect among other factors. In the past, the liquid dispersions have been preferred over the solid dispersions for new drug candidates in early formulation development as they reduce the development time and cost [4, 5]. However, with the development of new technologies for the preparation of solid dispersions and new excipients, this is going to change and both formulation types offer a lot of opportunities. For example, Kaletra® was launched as a soft gel liquid dispersion capsule (September 2000) but substituted by a solid dispersion tablet formulation (October 2005), because of higher stability and higher dose per unit. Regardless of dosage formulation (orals, parenterals, or others), the industry is also under the scrutiny of the agency because of the complexicity associated with safety, quality and efficacy of formulation dosages involving excipients, APIs, manufacturing and/or delivery technologies.

Figure 1 - An outline of complexity in drug development
Figure 1 illustrates the complexity of formulation development in highly regulated environments requiring multi-facet components to achieve a particular dosage with sound quality, safety and efficacy. It is not only the active ingredients (APIs) and excipients, but the manufacturing and/or the delivery technologies that play an equally important role in achieving the desired quality, safety and efficacy of a drug product.
The scope of this article is to cover the challenges and opportunities stemming from poorly soluble molecules for oral delivery, and how the industry is adjusting by adopting new nonconventional formulation technologies to bring new drugs into the market. The article will cover the biopharmaceutical classification systems (BCS) of drugs, and the factors that could lead to the increase of solubility and permeability of drugs, and an understanding of supersaturation, and the excipients’ role in development of liquid and solid dispersions.
Biopharmaceutical Classification Systems (BCS)
Table I shows the classification of drugs according to the current understanding of BCS.
The solubility and/or permeation enhancement of molecules for Class II, III and IV can be achieved to meet the so-called biopharmaceutical fi tness by many approaches as outlined in Figure 2 [6, 7].
Over 90% of the marketed drugs qualify under Class II and Class IV [8]. The continued trend of increased number of poorly soluble and permeable molecules is alarming and has brought the industry nearly to a standstill. The work continues to overcome these formulation challenges to fi nd the appropriate solutions for these poorly soluble and permeable molecules.
Table 1.

The solubility in water defined by activity coefficient and the crystal terms of a drug molecule and can be represented by Eq. 1 [9]:
Log Sw = 0.8 – log Kow - 0.01 (MP – 25)
where, Sw is the aqueous solubility of drug expressed in mol/L, Kow is the octanol/water partition coefficient, MP is the melting point in oC.
This article aims at examining the challenges in developing stable formulations and the role excipients play in maintaining the supersaturation of drugs in the dosages.
Role of Excipients in the Formulation Development
Liquid Dispersions

Figure 2 - Factors lead to increased solubility and permeability of Class II and IV, and Class III drugs.
Liquid dispersions have been the subject of continued interest in drug delivery and development [10]. The lipid-based delivery systems (LBDS) have been identified as “true liquid dispersions for self (micro) emulsification drug delivery systems “(S(M)EDDS)” and applied successfully in development of poorly soluble lipophilic molecules with 2 < logP > 4. SEDDS and SMEDDS provide an easy scale up for manufacturing of these dosages in oral solutions, liquid/semi-solid for soft gel, and/or pellets for hard gel capsules or tablets, and are amenable to those requiring highest achievable doses [8].
The excipients for LBDS are comprised of a wide range of molecular structures, compositions and functionalities. These include but are not limited to solubilizers/surfactants with polar and non-polar entities that are able to form self-assembled aggregates in aqueous solutions, and are able to maintain the desired concentrations in gastrointestinal (GI) fluids. Most importantly, these surface active excipients (natural or synthetic) are characterized by their critical micelle concentration (CMC) and/or hydrophilic lipophilic balance (HLB) values [11]. The surfactant molecules with higher molecular weight tend to possess the lower CMC values and vice versa. In contrast, those with higher hydrophilicity, possess the higher HLB values. For instance, KolliphorTM P 188 or Poloxamer 188 (MW 8000) and KolliphorTM P 407 or Poloxamer 407 (MW 12000), possess respectively, the CMC values of 1.4 x 10-3 M and 8.0 x 10-4 M, and the HLB values of 29 and 23. In addition, these surfactants/solubilizers meet the sound safety and regulatory standards, and are compatible with most of the drugs.
In the self-emulsifying process, the surface active molecules aggregate in particulates from the pre-concentrate in a suffi cient aqueous environment. Figure 3 illustrates the mechanism underlying the emulsifi cation involving the formation of small particulates from a clear, transparent pre-concentrate upon dilution for absorption through GI tract membrane. The key step requires maintaining the drug in solution (supersaturation) to facilitate the permeation and absorption. The digestive enzymes can precipitate the API in GI tract from micro/emulsions by lipolysis of fatty acid esters of the surfactants/solubilizers. Therefore, maintaining the drugs in the solutions is unequivocally important in the GI fl uids, but it could be challenging, especially, if the amounts of solubilizers are not sufficient. Therefore, the higher percentages of solubilizers are usually required for the stability, and yield smaller particulates for faster absorption through GI tract [12].
Figure 4 illustrates a pseudo-ternary phase diagram of surfactant, cosurfactant, and oil in the aqueous phase. It is apparent that increasing the amounts of surfactant and co-surfactants leads to smaller particles or micro or nano-emulsions or SMEDDS/SNEDDS as shown by the blue domains, while the lower amounts result in larger emulsions (as shown by the yellow domains). The typical particle sizes of the smaller particulates could range 50-200 nm, whereas, the larger particulates could have the size of about 400-500 nm.
The typical components of SEDDS include:
Surfactants (HLB >12) and co-surfactants (HLB<12)
Examples: Polyoxyl 35 castor oil (Kolliphor™ EL), Polyoxyl hydrogenated 40 castor oil (Kolliphor™ RH 40), Polyoxyl 15 hydroxystearate (Kolliphor™ HS15), Polysorbate 80, vitamin E-TPGS (Kolliphor™ TPGS) , Transcutol® P, Labrafi l®
1944 C, Kolliphor™ P 188 (Poloxamer 188), Kolliphor™ P407 (Poloxamer 407), and Kolliphor™ P 124 (Poloxamer 124) among others.
Triglycerides (Oils)
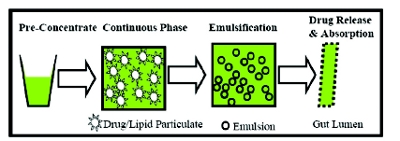
Figure 3 - A self-emulsifying process from the pre-concentrate
Medium chain triglycerides or MCT (C6-C12):
Glyceryl tricaprylate/caprate: Captex® 300; Miglyol® 810; Miglyol® 812; Neobee®M-5
Long Chain Triglycerides of LCT (C14-C18)
Corn oil, soybean oil, saffl ower oil, olive oil
Mono- and Di-glycerides:
Glyceryl caprylate/caprate (Capmul® MCM, Imwitor® 742), Glycerol Monocaprylate (Imwitor® 308, Glycerol monooleate(Capmul® GMO))

Figure 4 - pseudo-ternary phase diagram of emulsifi cation systems derived from surfactant-oil-water mixtures.
Propylene glycol esters:
Propylene glycol monocaprylate (Capmul® PG-8), Propylene glycol monolaurate (Capmul® PG-12, Lauroglycol®)
Co-solvents/fatty acids:
Propylene glycol, ethanol, polyethylene glycols (PEG 300, 400, 600), oleic acid, palmitic acid, stearic acid, linoleic acid, linolenic acid.
Examples
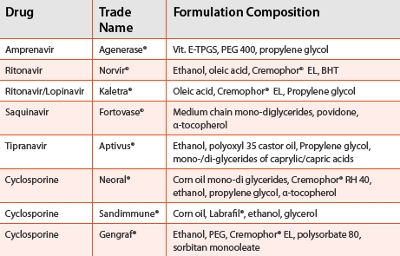
These lipid-based products have been approved in oral and parenteral formulation dosages due to their remarkable solubilization and biocompatibility with superb tolerability and safety profi les in animals and humans. There are innumerable examples and case studies involving self-emulsifying systems for in vitro and in vivo performances of model drugs [13]. It is beyond the scope of this article to cover all of them, but a few examples are highlighted. For instance, Solutol® HS15 (rebranded to Kolliphor™ HS15) [14], showed a signifi cant improvement in the bioavailability of vinpocetin in self-emulsifying system as compared to tablet formulation [15]. In another study, Kolliphor™ RH40 (Cremophor® RH 40) in a self-emulsifying system signifi cantly improved the bioavailability of atorvastatin in beagle dogs than the conventional tablets [16]. In general, the larger amounts of surfactants in formulations are critical for yielding the smaller particles for enhanced drug permeation and absorption [17]. The liquid dispersions have been primarily limited only to soft gel or hard gel liquid fills, but current studies suggest that these formulations can be improved to develop self-emulsifying tablets. In a recent study, the authors have shown that a waxy Kolliphor™ HS15 can be used to design diazepam formulation in microcrystalline cellulose pellets with similar release profiles to conventional tablets [18]. In another study, a Neusilin® based solid emulsifying system was developed for tableting involving Kolliphor™ EL (Cremophor® EL) as a surfactant for better stability and performance than the liquid dispersion [19]. Cuine et al. studied effects of fatty acid compositions on the lipolysis and observed that the saturated fatty acids in Kolliphor™ RH 40 were less susceptible to digestive enzymes as compared to unsaturated Kolliphor™ EL and Polysorbate 80 or short chain Labrosol® [20]. As a result, the long saturated fatty acids of Kolliphor™ RH 40 prevented the precipitation of danazol and increased its bioavailability in the beagle dogs.
The systemic absorption of drugs occurs through portal vein and intestinal lymphatic cells. A plausible mechanism of GI absorption from the lipidbased systems remains elusive. The ability to predict bioavailability, lymphatic transport and uptake, first pass hepatic metabolism, and food eff ects are subjects of continued interest and investigation. It is understood, however, that bypassing the liver reduces the first pass liver metabolism and improves the intestinal absorption [21]. Thus, primary interest is to avoid the hepatic fi rst-pass metabolisms or “clearance” in order to enhance the oral absorption of lipid particulates via portal vein and intestinal lymphatic routes [22, 23]. Interestingly, the lymphatic transport is higher with long-chain triglycerides than medium chain triglycerides [24]. Though the exact mechanism of lymphatic transport is not fully understood but it is believed that lipoproteins are involved in lymphatic transport of lipophilic drugs. Figure 5 illustrates the transport through the portal vein, which is a key pathway for the absorption of low molecular weight drugs to enter systemic circulation because of the higher fl uid fl ow rate (>500-fold) compared to lymphatic flow [21].
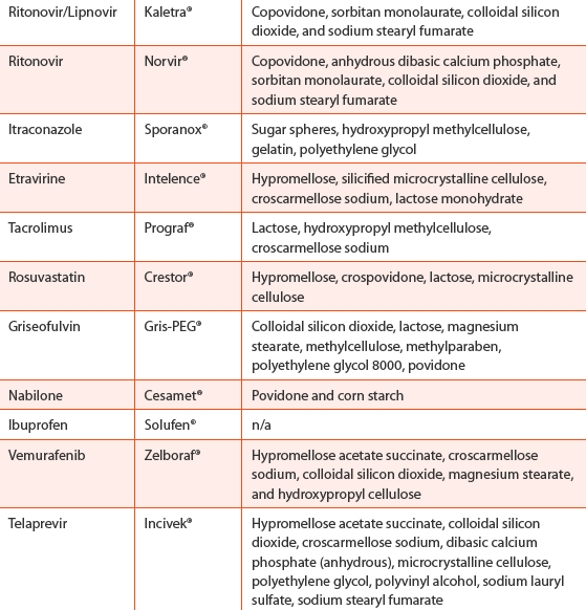
A number of commercial products have been launched over the years with lipid-based micro/emulsions, micellar suspensions, oil solutions and selfemulsification systems (SES). Table 2 lists some of marketed products [25].
Solid Dispersions
Solid oral dosages formulations (SODF) have continued to be the focus in the industry. Especially, the solid dispersions are fully exploited for highly crystalline, high melting lipophilic drugs, wherein, the drugs are converted to a high energy amorphous powder to increase the solubility and bioavailability [26]. Figure 6 illustrates a number of solid dispersion technologies well developed (e.g. hot melt extrusion, spray drying, coprecipitation, melt granulation, lyophilization), and in the early stages of development (e.g. Kinetisol®, microwave irradiation, electrospinning) [27- 29]. This article will not dwell on every technology but instead will focus on the amorphous solid dispersions in general, and lay out the role of excipients in the formulation development.
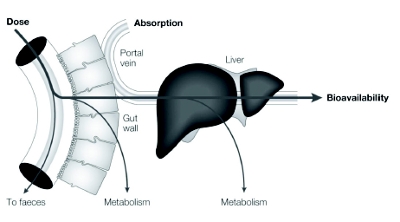
Figure 5 - Pathways leading to increased absorption and bioavailability
Enablers are the excipients that play a crucial role for a robust formulation. The structure-function hallmarks of excipients, interaction with drugs, and their consequences on long- term stability and formulation performance, are considered for compatibility and predicting the stability of amorphous dispersions [30].
The commonly used excipients for solid dispersions are:
Neutral cellulose derivatives
Hydroxypropyl methylcellulose (Hypromellose): HPMC
Hydroxypropyl cellulose: HPC
Cellulose acetate butyrate: CAB
Examples of Lipid-Based Approved Drugs
Acidic cellulose derivatives
HPMC acetate succinate: HPMC-AS
HPMC phthalate: HPMC-P
Cellulose acetate phthalate: CAP
Neutral (non-cellulosics)
PVP: Polyvinylpyrrolidone (K12, K17, K25, K-30, K-90)
Copovidone: Vinylpyrrolidone - vinyl acetate copolymer
Soluplus®: PEG-co-PVAc-co-PVCap
Kollicoat® IR: PEG-co-PVA
Polyethylene oxide: PEO (MW > 20K) or PEG (MW < 20K), PEG/PPG
Ionic (non-cellulosic)
Methacrylic or acrylic copolymers: Kollicoat® MAE 100 P, Eudragit® EPO, L100-55, RS, RL.
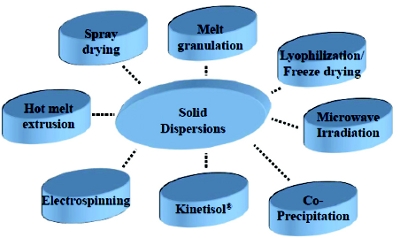
Figure 6 - Methods for preparation of solid dispersions
The polymers for solid dispersions have been studied and a large body of literature exists [31, 32]. Given the vast scope of formulation topic, this article will focus on commonly used polymers in the amorphous solid dispersion (ASD), especially, those used in hot melt extrusion for solid solutions characterized by complete miscibility of drugs with the polymer matrix with one combined glass transition temperature (Tg) of a polymer and drug [33, 34]. Though a number of hydrophilic polymers including polyvinylpyrrolidone (PVP), copovidone (VP-VA copolymer), hydroxylpropyl methylcellulose (HPMC), hydroxypropyl methylcellulose acetate succinate (HPMCAS), hydroxypropyl cellulose (HPC), and polymethacrylic acid copolymers among others are available, only a few are suited for melt extrusion because of their higher Tgs, impediment in processing, and instability to withstand high temperatures during the process [35]. Soluplus®, a novel graft copolymer consisting of polyethyleneglycol as a backbone and vinyl acetate-vinylcaprolactame side chains, is thermally stable and can withstand the temperatures as high as 220oC without degradation. Table 3 lists the Tg and degradation temperature (Tdeg) of some of the polymers that will help guide the melt extrusion process [36].
Figure 7 illustrates the process of melt extrusion where the crystalline drug is dissolved in the polymer on melting when heated typically to temperatures at > 20 - 40 oC above the Tg of the polymer, resulting in formation of a clear glassy solid solution or extrudates. The extrudate can be further pelletized, blended with other fi llers and disintegrants, and compressed into tablets.

Figure 7 - Illustration of solid solutions by hot melt extrusion of a polymer and crystalline API
As indicated, many of the polymers can be used in solid dispersion processes that do not require higher temperatures such as spray drying [37] and electro-spinning [29a]. But, those are limited because they are thermally labile, and will decompose or degrade on exposure to higher temperatures. Soluplus® was especially designed for preparation of solid solutions [38]. In earlier studies, the solid solutions of Soluplus® significantly increased the solubility and bioavailability of several model drugs examined [39,40]. Soluplus® is accessible to many of poorly soluble compounds which otherwise had the least opportunities for development because of its unique solubilization characteristics.
Understanding the Supersaturation Phenomenon
The thermodynamic stability of amorphous solid dispersions (ASD) has been investigated quite often [41]. A rapid crystallization and when no rapid crystallization occurs predicting the stability of a metastable amorphous dosage can be challenging [30]. Figure 8 illustrates the processes that lead to nucleation/crystallization of an API from ASD on standing in the storage conditions, under in vitro dissolution, or in the GI fl uids. As the nucleation commences, no supersaturation can be achieved, and condition as shown in Figure C will prevail, leading to a thermodynamically less stable system. If the crystallization is slower than the dissolution rate, the supersaturation can be achieved as shown in Figure A, leading to a kinetically stable system. The slower dissolution rate may indicate the start of nucleation faster than dissolution rate, resulting in the condition in Figure B [39]. The polymeric carriers can infl uence the conversion of amorphous to crystalline drug, which may impede or accelerate the dissolution. The variability in the dissolution rates is infl uenced by (i) nature of polymers, (ii) polymer- API interactions, and (iii) moisture uptake of composite blends. Thus, to attain the supersaturation, less hygroscopic polymers with abilities to solubilization and complexation are crucial factors for increasing the solubility and bioavailability of drugs.
Table 3. Tg and Tdeg of different polymers*
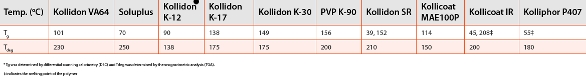
Figure 9 illustrates the hypothetical in vitro solubility profi les of a drug from amorphous solid dispersions. When applied in the context of thermodynamically stable ASD, the curve A simulating the conditions in Figure 8A, will prevail, representing a “parachute” type of behavior yielding much longer release than those dispersions in B and C states. Thus, curves B and C will simulate the situation in Figure 8B and 8C, respectively, where an onset of nucleation will crystallize the drug much sooner than the curve A. The nucleation followed by crystallization processes (B and C) ceases the opportunity to fully attain the supersaturated state. Collectively, it can be suggested that the thermodynamic stability of an ASD is as important as the kinetic stability of powder in the aqueous solution for supersaturation.
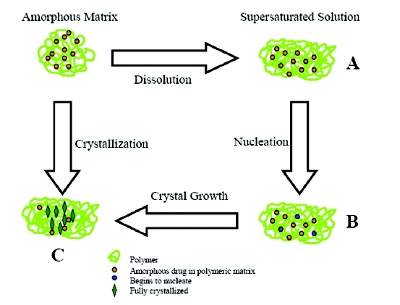
Figure 8 - Illustration of supersaturation, nucleation, and crystallization phenomena
Examples
The interactions and complexing abilities of polymers are critical for enhancing solubility and maintaining supersaturation. The polymers with inherent binding sites resulting from the functional moieties enable the hydrogen-bonding and/or Van der Waal’s interaction with drug molecules, leading to API’s stability in the matrix [42]. The higher drug loading may lead to precipitation or crystallization because of lack of suffi cient interaction sites on the polymers. Interestingly, polymers bearing amide (N-C=O) linkages have tendencies to form complexes with actives more readily than those with alcohol (C-OH) moieties. For examples, polyvinylpyrrolidone (PVP), Vcopovidone (Kollidon® VA64) and Soluplus® are better solubilizers than those alcohol-based cellulosic polymers: hydroxypropyl cellulose (HPC), hydroxypropyl methylcellulose (HPMC) or hydroxypropyl methylcellulose acetate succinate (HPMCAS) and polyethyleneglycol-polyvinylalcohol (Kollicoat® IR) polymer. Many polar organic solvents with the amide linkages such as 2-pyrrolidone, N-methyl pyrrolidone, formamide, acetamide) are better solvents with signifi cantly greater solubilization characteristics than those with hydroxyl or ketone groups (e.g. alcohols and acetone) [36].

Figure 9 - Illustration of in vitro release profi les of a drug under certain plausible conditions
Intermolecular interactions in the amorphous dispersions are as important as the drug loading. It often leads to drug nucleation/crystallization on storage or precipitation in the dissolution media if the drug loading exceeds the saturation threshold. The stability is also dependent upon the lengths and molecular weight of the polymers; a higher molecular weight leads to signifi cantly greater entanglement with drug molecules. In homopolymers like PVP K-12 and PVP K-30, the stronger interactions will last longer in K-30 than in small K-12 polymer. Polymers like Soluplus® with an average molecular weight of 118,000 and multiple binding sites on the lipophilic residues, created by grafting vinyl acetate and vinylcaprolactame moieties, also provide stronger entanglement than those lacking such moieties or interaction sites [36]. These prerequisites with phenomenal complexing and solubilization behaviors of Soluplus® are critical for maintaining the supersaturation, as illustrated in Figure 10. The stronger interactions may also lead to slower release or slower API’s precipitation and/or degradation. In such cases, it is inevitably important to assess the other factors such as excipient-API compatibility, drug loading, and hygroscopicity and degradation of polymers and drugs for a stable and robust formulation.
The stronger interaction with drugs leads to stabilization and prevention of crystallization, and thus, faster release of drug. In some instance, that can also lead to dissolution lag time depending upon the drug molecules. In a recent study, Soluplus® showed the faster release of a high melting compound than those of cellulose acetate phthalate (CAP) and Kollidon® VA64 dispersions [43]. It has been shown that Kollidon® VA64 with 0.2% Poloxamer 188 led to far less degradation (0.2% vs. 45%) of a high melting API in neutral polymers as compared to anionic polymers Eudragit® 100- 55 and HPMCAS extrudates [44]. In some cases, the binary polymers have shown to stabilize the dispersions better than a single polymer [45]. These authors have shown that Eudragit® EPO resulted in a decrease of itraconazole release as the dissolution continued over an hour (kineticallyunstable), but the addition of 10-20% Kollidon® VA64, helped stabilize the binary dispersions.
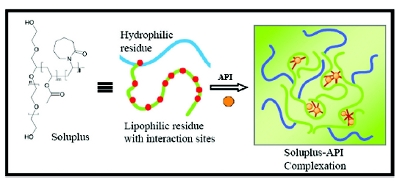
Figure 10 - Complexing ability of Soluplus® with an API
The dissolution rate also depends on the rate of hydration of the polymer. For instance, fenofi brate dispersions with Kollidon® VA64 showed quicker release than Soluplus®, presumably due to faster hydration of Kollidon® VA64 with respect to Soluplus®, as shown in Figure 11. When compared with PVP K-12, the fenofi brate amount increased in the fi rst 15 min of dissolution, and then decreased as the dissolution continued. These changes in dissolution profi les might be attributed to precipitation of drug due to lower molecular weight of the polymer and hence a lesser degree of entanglement with the polymeric chain [34]. The HPC dispersions however did not release fenofi brate due to gelling of the polymer and immediate precipitation. Tsinman et al. have demonstrated that the Soluplus® maintains the supersaturation over 16 hours from carbamazepine dispersions at pH 1.2 and FaSSIF conditions [46]. In a recent study it was found that PVPs, Kollidon® VA64 and Soluplus® were more eff ective crystallization inhibitors for fenofi brate than cellulosic polymers [47].

Figure 11 - Dissolution profi le of fenofi brate from solid dispersions of diff erent polymers
Conclusions
This article reviews the challenges in the formulation, and the ways the industry currently perceive these opportunities for drug development. With a continued trend in the number of poorly soluble new chemical entities (NCEs), and increasing time and costs in development, the industry has begun evaluating all options to streamline development strategies. As a result, the surfactant or lipid-based self-emulsifying delivery systems (SEDDS/SMEDDS) and polymer-based solid dispersions/solutions have become the viable formulation options in the drug development. This review also describes how the excipients play important roles in both liquid and solid dispersions.
Table 4. Marketed Solid Dispersions Products
The choices between the two dosages and with desire to market the drugs faster, the self-emulsifying liquid dispersions have been preferred in the past over solid dispersions. The lack of viable polymers for solid dispersion technology, thermal instability of APIs, and the stringent regulations on residual solvents have all contributed to the growth of lipid-based liquid dispersions. However, the situation is changing and solid dispersions are gaining ground due to new manufacturing technologies, e.g. hot-melt extrusion, and also new excipients especially designed for solid dispersions such as Soluplus®.
The challenges are still enormous, and there is no magic formulation or process for developing all drug compounds. Though both the technologies have been fully developed and practiced for over 2 decades, only a handful of drugs have been marketed by these technologies , as shown in Table 4. With advent of new excipients, the industry will have better choices to select one process over the other in order to create better medicines for tomorrow.
References
- C. Giliyar, D. T. Fikstad, S. Tyavanagimatt, “Challenges and opportunities in oral delivery of poorly water-soluble drugs”. Drug Delivery Technol., 2006, 6, 57-63
- J. Breitenbach, “Melt extrusion: from process to drug delivery technology”, Eur. J. Pharm. Biopharm., 2002, 54, 107-117. 3
- D. J. Hauss, “Oral lipid-based formulations: enhancing he bioavailability of poorly water soluble drugs”, volume 170, Informa Healthcare.
- P. P. Constantinides, “Enabling Oral Drug Delivery with Lipids: Trends and Perspectives”. AAPS Workshop, Lipid‐based delivery for improving drug absorption- Mechanistic Understanding and practical approaches, Baltimore, USA, 23-24 April 2012.
- Li, P., Zhaob, L., Developing early formulations: Practice and perspective, Int. J. Pharm., 2007, 341, 1-19.
- Mehta M. AAPS/FDA Workshop on bio-pharmaceutics classification system (2002).
- J. Alsenz, M. Kansy, “High throughput solubility measurement in drug discovery and development”, Adv. Drug Del. Rev., 2007; 59: 546-567.
- B. Griffin, Advances in lipid-based formulations: Overcoming the challenges of low bioavailability for poorly water soluble drug compounds, Am. Pharm. Rev., March 2012, 41-47.
- S. H. Yalkowsky, Solubility and Solubilization in Aqueous Media, Oxford University Press, NY (1999), p. 67.
- S. S. Ren, B. D. Anderson, “What determines drugs solubility in lipid vehicles: Is it predictable?” Adv. Drug Del. Rev., 2008; 60, 638-656.
- T. Reintjes, “Solubility enhancement with BASF pharma polymers: Solubilizer compendium”. BASF SE Pharma Ingredients & Services, Germany, October 2011.
- de Smidt, C. P., Campanero, M. A., Trocóniz, I. F., “Intestinal absorption of penclomedine from lipid vehicles in the conscious rat: contribution of emulsification versus digestibility”, Int. J. Pharm., 2004, 270, 109-118.
- Mullertz, A., Ogbonna, A., Ren, S., Rades, T., New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs, J. Pharm. Pharmacol., 2010, 62, 1622-1636.
- S. Ali, T. Rosen, “Rebranding of BASF Solubilizers”, Tablets & Capsules, April 2012.
- S. X. Cui, S. F. Nie, L. Li, C. G. Wang, W. S. Pan, J. P. Sun, “Preparation and evaluation of self-microemulsifying drug delivery system containing vinpocetine”, Drug Dev. Ind. Pharm. 2009, 35, 603-11.
- Shen, H.R., Zhong, M.K., Preparation and evaluation of self microemulsifying drug delivery systems (SMEDDS) containing atorvastatin., J. Pharm. Pharmacol. 2006, 58, 1183–1191.
- Z-G. Gao, H-G. Choi, H-J. Shin, K-M. Park, S-J. Lim, C-K. Kim., “Physicochemical characterization and evaluation of a microemulsion system for oral delivery of cyclosporin A”, Int. J. Pharm., 1998, 161, 75–86.
- Abdalla A, Mader K. “Preparation and characterization of a self emulsifying pellets formulation”, Eur. J. Pharm Biopharm. 2007, 66: 220‐226.
- S. Gumaste, D. Dalrymple, A. Serajuddin, “Development of lipid-based self-emulsifying tablets with Neusilin® US2: Design consideration”. AAPS Northeast Regional Discussion Group, Rocky Hill, CT (April 2012).
- J. F. Cuine, J., C. L. McEvoy, W. N. Charman, C. W. Pouton, G. A. Edwards, H. Benameur, C. J. H. Porter, Evaluation of the impact of surfactant digestion on the bioavailability of danazol after oral administration of lipidic self-emulsifying formulations to dogs, J. Pharm. Sci., 2008, 97, 995-1022.
- C. J. H. Porter, N. L. Trevaskis, W. N. Charman, “Lipids and lipid-based formulations: Optimizing the oral delivery of lipophilic drugs.” Nature Reviews Drug Discovery 2007, 6, 231–248.
- Y. Jiao, N. Ubrich, M. Marchand-Arvier, C. Vigneron, M. Hoffman, T. Lecompte, and P. Maincent. In vitro and in vivo evaluation of oral heparin-loaded polymeric nanoparticles in rabbits, Circulation 105:230Y235 (2002).
- M. Aprahamian, C. Michel, W. Humbert, J. P. Devissaguet, and C. Damge. Transmucosal passage of polyalkylcyanoacrylate nanocapsules as a new drug carrier in the small intestine. Biol. Cell 61:69Y76 (1987).
- S. M. Caliph, W. N. Charman, and C. J. Porter. “Effect of short-, medium-, and long-chain fatty acidbased vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymphcannulated and non-cannulated rats”, J. Pharm. Sci. 2000, 89: 1073Y1084 .
- Fatouros, D. G., Karpf, D. M., Nielsen, F. S., Mullertz, A., Clinical studies with orl lipid-based formulations of poorly soluble compounds, Therapeut. Clin. Risk Manag., 2007, 3, 591-604.
- B. E. Padden, J. M. Miller, T. Robbins, P. D. Zocharski, L. Prasad, J. K. Spence, J. LaFountaine, “Amorphous solid dispersions as enabling formulation for discovery and early development”, Am. Pharma. Rev., 2011, Jan/Feb. 66-73.
- J. C. DiNunzio, C. Brough, D. A. Miller, R. O. Williams III, J. W. McGinity, “Applications of KinetiSol® dispersing for the production of plasticizer free amorphous solid dispersions”, Eur. J. Pharm. Sci. 2010, 40, 179–187
- M. Moneghini, N. De Zordi, D. Solinas, S. Macchiavelli, F. Princivalle, “Characterization of solid dispersions of itraconazole and vitamin E TPGS prepared by microwave technology”, Future Med. Chem. 2010, 2, 237–246.
- (a) S. Ali, N. Langley, D. Djuric, K. Kolter, S. Mirza, “Electrospinning for Solid Dispersions of Poorly Soluble Drugs”, CRS 2011; (b) Z. K. Nagy, A. Balogh, B. Vajna, A. Farkas, G. Patyi, A. Kramarics, G. Marosi, “ Comparison of electrospun and extruded Soluplus® -based solid dosage forms of improved dissolution”, J. Pharm. Sci., 2011, 1-14 (DOI 10.1002/jps.22731)
- K. C. Waterman, “Accelerated stability assessment program (ASAP): Using science to set shelf life”, Pharm. Outsourcing, 2012, 40-46.
- K. Dhirendra. S. Lewis, N. Udupa, K. Atin, “Solid dispersions: A Review, Pak. J. Pharm. Sci., 2009, 22, 234-246.
- Y. S. R. Krishnaiah, “Pharmaceutical technologies for enhancing oral bioavailability of poorly soluble drugs”. J Bioequiv Availab., 2010, 2, 28- 36.
- F. Qian, J. Huang, Q. Zhu, R. Haddadin, J. Gawal, R. Garmise, M. Hussain, “Is a distinctive singe Tg a reliable indicator for the homogeneity of amorphous solid dispersion?, Int. J. Pharm., 2010, 395, 232-235.
- M. A. Repka, “Hot melt extrusion”, Am. Pharm. Rev. 2009, 18-27.
- C. A. McKelvey, “Design of stable extruded solid dispersions”. CRS July 9, 2010, Educational Workshop 2, Portland, Oregon.
- K. Kolter, M. Karl, S. Nalawade, N. Rottmann, „Hot melt extrusion with BASF pharma polymers: Extrusion Compendium, Oct. 2010.
- D. Smithey, J. Fennewald, J. Gautschi, M. Crew, S. Ali, Y. Lan, and N. Langley, “Evaluation of the Polymer Soluplus® for Spray-Dried Dispersions of Poorly Soluble Compounds”, AAPS 2010.
- Soluplus®- Technical Brochure, BASF 2009.
- S. Ali, N. Langley, D. Djuric, K. Kolter, “Soluplus- a novel polymer for hot melt extrusion, Tablets & Capsules, October 2010.
- H. Hardung, D. Djuric, S. Ali, “Combining HME and Solubilization: Soluplus® - The Solid Solution”, Drug Del. Tech., 2010.
- B. C. Hancock, M. Park, “What is the true solubility advantage of the different forms”, Pharm. Res. 2000, 17, 397-404.
- D. E. Alonzo, G. G. Z. Zhang, D. Zhou,Y. Gao, L. S. Taylor, “Understanding the behavior of amorphous pharmaceutical systems during dissolution”, 2010, 27, 608-618.
- J. C. DeNunzio, “Melt Extrusion: Technology Overview and Applications for Early Stage Formulation Development”, IPRIME Workshop, University of Minnesota, Jan. 2012.
- D. A. Miller, “APV/AAPS conference on hot melt extrusion”, April 2011.
- K. Six, G. Verreck, J. Peeters, M. Brewster, G. V. D. Mooter, “Increased physical stability and improved dissolution properties of itraconazole, a Class II drug, by solid dispersions that combine fast- and slow-dissolving polymers”, J. Pharm. Sci., 2004, 93, 124–131.
- K. Tsinman, O. Tsinman, N. Langley, and S. Ali, “Soluplus® Maintains the Supersaturation of Carbamazepine from Amorphous Solid Solutions/Dispersions.” AAPS (2012).
- M. A. Repka, personal communication.
Dr. Shaukat Ali has more than 18 years of experience in the pharmaceutical industry including 8 years at BASF, where he is supporting the solubilization platform. He received his Ph.D. in Chemistry from the City University of New York and pursued his postdoctoral work at the University of Minnesota and Cornell University. Dr. Ali is an adjunct faculty at the College of Pharmacy of University of South Florida. Dr. Ali’s areas of expertise include drug solubilization, liposome drug delivery, controlled release and film development technologies. Dr. Ali is a member of the editorial boards of American Pharmacetical Review, Contract Pharma, Drug Delivery & Development and International Journal of Pharmaceutical Investigation. He is also USP panel of experts for General Chapters-Physical Analysis. He has authored over 25 scientific articles and is coinventor in 14 US patents.
Dr. Karl Kolter started his career in the pharmaceutical development department of what was formerly Knoll AG, Ludwigshafen, in 1986 where he was involved in the development of oral liquids, parenterals, sustained release drug delivery systems and drugs made by direct compression. In 1993, Dr. Kolter joined BASF AG, where he was responsible for R&D in pharmaceutical excipients, drug formulations and the application technology of vitamins and carotenoids for pharma and food. Dr. Kolter’s current work is the development of innovative excipients mainly for solid oral dosage forms, which has already resulted in various new products in the Kollicoat and Kollidon range. Dr. Kolter obtained his Ph.D. in pharmaceutical chemistry at the University of Mainz (Germany). He has published more than 100 articles and posters, and inventor in 40 patents.