Introduction
A placebo-controlled study is a means of testing a drug for safety and efficacy in a group of subjects that receive the treatment. Current placebo identity tests typically utilize an HPLC identity method for the active compound to confirm the absence of the active (i.e., negative identity). In this review, the development and application of transmission Raman spectroscopy (TRS) with chemometric modeling for positive placebo identification testing will be applied to drug products, illustrated for several compounds and their respective placebos.
Placebo identification tests are a clinical manufacturing requirement, and when implemented in a negative mode, data are evaluated against the specification “There is no active detected”. Clinical placebos share the same physical appearance as the active tablets or capsules, as required for blinded studies, hence definitive identification of both the active and placebo (absence of active) are necessary release tests. Spectroscopic test methods utilizing a standard library provide a robust approach for evaluating the chemical identity of both placebos and actives. In addition, spectral testing using chemometric models can compare placebo results against a database of numerous placebos and active drugs instead of assaying for a single active ingredient. Spectroscopic methods utilizing rapid, chemometric-based spectroscopic technologies create efficiencies and minimize workload due to minimal sample preparation and automated data analysis. Finally, chemometric models provide benefits over traditional spectral comparisons such as eliminating a need for storage and maintenance of reference standards (e.g., API, tablets and capsules) and reduce subjectivity in determining if sample data “compares favorably to a reference standard” or demonstrates “no active XYZ present,” especially for complex drug excipient matrices.
Experimental
For the experiments reported here, a Cobalt transmission Raman instrument (TRS100) was used with the following settings:
- Laser Power: 0.65W
- Exposure: 0.5 sec.
- Accumulations: 180
- Detector: CCD
- Read Optics: Small
- Laser Spot Diameter: 2mm
- Scan Range: 40 – 2400 cm-1
- Run Time: 90 seconds
Samples tested included 12 unique active drugs and their placebos, covering a total of 34 different strengths for tablets and capsules. Data analysis was performed using the chemometrics software package Solo (Eigenvector Research, Inc.).
Results and Discussion
Drug product placebos are made to match the active product physical properties such as color, shape and size in order to facilitate blinded clinical studies. However, in some cases, the manufactured placebo may be prepared using a standard formulation that differs from the active drug product due to not only the absence of the active compound, but also with respect to the use of other raw materials and excipients. In other cases, the placebo formulation may be identical to the active formulation with the obvious exception of absence of the active drug.
In this study, the model used included both tablets and capsules which also have different unit formulas. In summary, the completed positive placebo identification model used contained a library of products and placebos which varied in formulation, shape, size, color, and presentation (e.g., capsule versus tablets).
Positive placebo identification tests were executed by initially creating a library of data from 12 active and placebo tablets and capsules covering 34 different dosage strengths ranging from 0.5 mg (0.25% drug load) to 300 mg (30% drug load). The initial model included library items of both tablets and capsule placebo formulations that differed from the active formulations. Once created, the model was challenged with new lots of the same products and placebos, and the model was later challenged by addition of two products and placebos sharing the same unit formula.
The initial examination included principal component analysis (PCA) that evaluated the full Raman spectral range to determine if a difference could be observed between the different samples types (e.g., placebos, actives, tablets, capsules). Limited selectivity was observed; however, improvement was obtained when spectral ranges were subsequently narrowed to include regions containing active peaks from the products and no or very few peaks present for the placebos (Figure 1). In most cases, the products containing actives consistently produced peaks in the selected regions of 650 – 840 cm-1 and 1480 to 1780 cm-1 which are consistent with compounds containing aromatic rings, carbonyls and amines. Excipients may also have responses in these regions; however, they did not prove to have significant spectral contributions (Figure 2). Excipients showing peaks in this region include lactose and starch.
 Figure 1 - Spectral ranges were selected based on the presence of active peaks in regions where no placebo peaks were observed. Typically these peaks were due to aromatic, carbonyl and/or amine stretches.
Figure 1 - Spectral ranges were selected based on the presence of active peaks in regions where no placebo peaks were observed. Typically these peaks were due to aromatic, carbonyl and/or amine stretches.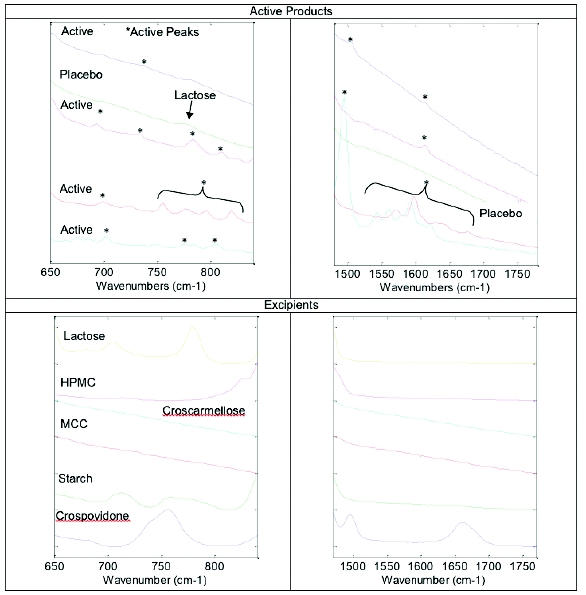 Figure 2 - Spectral regions demonstrating active and excipient peaks
Figure 2 - Spectral regions demonstrating active and excipient peaksReducing the spectral region to 650 – 840 cm-1 and 1480 to 1780 cm-1 demonstrated significant separation of actives and placebos; however, PCA analysis did not show complete discrimination as one active sample was present in the placebo “cluster” (Figure 3). Further evaluation showed this product to contain a low level active (2.5 mg), and similar formulation components to the placebos making discrimination challenging.
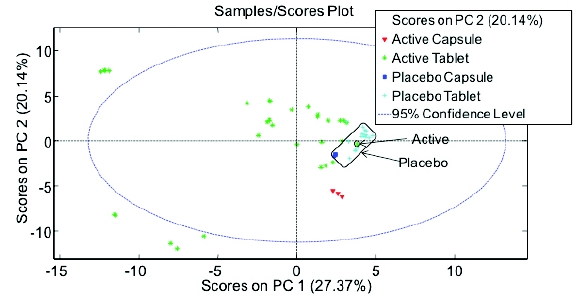 Figure 3 - PCA analysis; Preprocessing: 2nd Derivative (Filter Width = 15, 2nd order polynomial), SNV, Mean Center at ranges of 650 – 840 cm-1 and 1480 to 1780 cm-1
Figure 3 - PCA analysis; Preprocessing: 2nd Derivative (Filter Width = 15, 2nd order polynomial), SNV, Mean Center at ranges of 650 – 840 cm-1 and 1480 to 1780 cm-1Additional work explored using partial least squares discriminant analysis (PLSDA). Using the same spectral range and pre-processing parameters, PLSDA separated all placebos and actives (Figure 4). As demonstrated in the plot, the 95% confidence ellipse of the actives and placebos are clearly separated along principle component (LV 1).
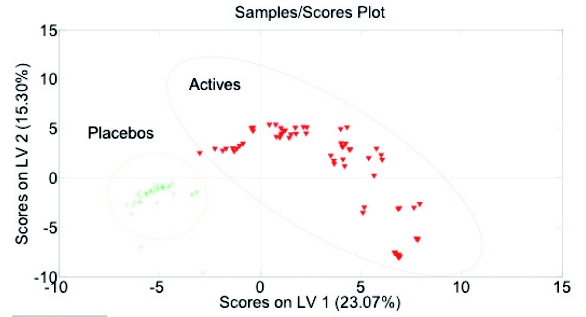 Figure 4 - PLSDA; Preprocessing: 2nd Derivative (Filter Width = 15, 2nd order polynomial), SNV, Mean Center at ranges of 650 – 840 cm-1 and 1480 to 1780 cm-1
Figure 4 - PLSDA; Preprocessing: 2nd Derivative (Filter Width = 15, 2nd order polynomial), SNV, Mean Center at ranges of 650 – 840 cm-1 and 1480 to 1780 cm-1The completed model included approximately 56 calibration active and placebo samples which was then validated with new samples (Figure 5). The validated model successfully classified the placebo and actives.
 Figure 5 - Calibration and validation results of model – Y-axis shows the model-estimated probability that a given sample belongs to the Placebo class.
Figure 5 - Calibration and validation results of model – Y-axis shows the model-estimated probability that a given sample belongs to the Placebo class.Once the model calibration and validation were complete, the validation data were combined with the calibration data and an independent sample set including replicates over multiple days (> 300 sample runs) was tested against the model. Without fail, the model accurately classified the active and placebo capsules and tablets (Figure 6 & Figure 7). As demonstrated, the actives and placebos were accurately classified, demonstrating reproducible results across multiple products, for both tablets and capsules. In addition, as shown in (Figure 6), 95% confidence ellipses demonstrated that the placebo and active separation were within the error of the model.
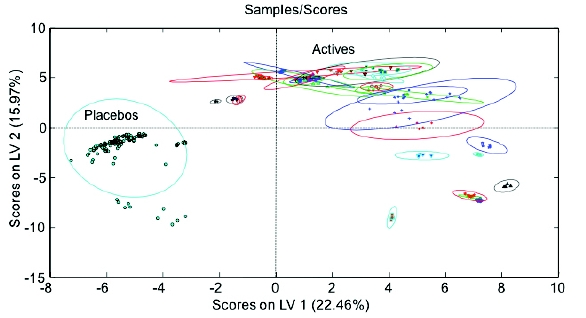 Figure 6 - Predicted Sample Results – scores on LVs 1 & 2
Figure 6 - Predicted Sample Results – scores on LVs 1 & 2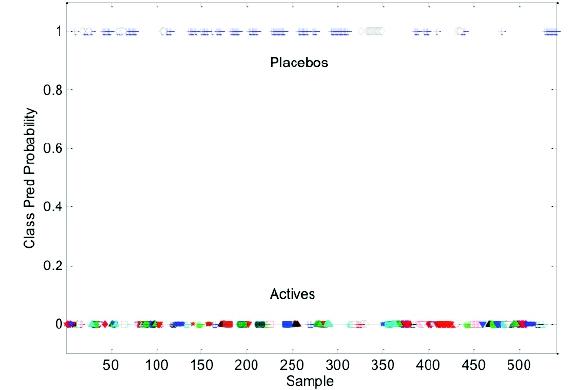 Figure 7 - Sample Test Results – Classified Placebo’s and Actives
Figure 7 - Sample Test Results – Classified Placebo’s and ActivesFinally, model robustness was challenged with the addition of two new products. In this case, the added products and placebos shared the same unit formulation, and the placebo formulations were significantly different from the placebos included in the model. The model was unable to distinguish the placebos from their respective actives (Figure 8), and classified the placebos as actives. The unit formula for the new placebos included an excipient spectral peak at approximately 790 cm-1 that was not consistent with the placebos included in the model and consequently, the excipient peak was interpreted as an active by the model (Figure 9). Further refinement of the model by removing the excipient spectral peaks associated with the new samples was not successful in accurately modeling the new data. Other model preprocessing parameters were also introduced (baseline correction, MSC, etc) without success.
 Figure 8 - Addition of new actives/placebos with similar formulations not separated
Figure 8 - Addition of new actives/placebos with similar formulations not separated  Figure 9 - Spectra of new active products with their corresponding placebo
Figure 9 - Spectra of new active products with their corresponding placeboBecause the model was not successful at properly classifying the new materials, a different modeling approach was required. Based in part on some earlier success1 with hierarchical models, it was decided to investigate this approach.
Hierarchical, or layered, models can be used to simplify a classification problem that is too complex to accurately resolve with simple standard linear models. Rather than attempt to separate all classes at once using a single model (one versus many), the problem is broken down into multiple steps. At each step, simple models are used to identify and eliminate either obviously-different classes, or to separate the possibilities into natural groups of classes. Subsequent steps (branches) use additional models to separate the remaining class possibilities further until all classes have been separated. By eliminating possible classes at each step, the requirements for subsequent models are simplified, thereby improving accuracy.
For the problem at hand, we were not able to successfully achieve the requisite level of discrimination between actives and placebos with a single model once the two new products were introduced – even when the new products were made part of the calibration set. This can be interpreted as a result of the new products introducing new sources of variance preventing the achievement of class discrimination with a single hyperplane. The earlier attempt to do so was too ambitious, at least with the tools at hand. It was thus necessary to break down the problem into smaller parts to eventually reach the modeling objective.
Taking a step back and looking at the PCA results for just the placebos – all of the original products plus new products (designated as NP1 and NP2), the directions defined by PCs 2 and 3 did a reasonable job of clustering this subset (Figure 10).
 Figure 10 - Scores for PC 3 vs. PC 2 - PCA model for placebos only
Figure 10 - Scores for PC 3 vs. PC 2 - PCA model for placebos onlyPreprocessing of the data has the potential to enhance separation by minimizing within-class variance and/or maximizing the distance between classes. The use of generalized least squares weighting (GLSW) collapses the three clusters for the original placebos into a single tight group, and spreads the NP1 and NP2 placebo groups much further apart (Figure 11) [1]. The addition of GLSW to the preprocessing recipe was insufficient to completely separate all placebos from all actives with the addition of the new products. However, the discrimination enhancement afforded by GLSW provides the impetus for creating the following modeling strategy, which was ultimately successful:
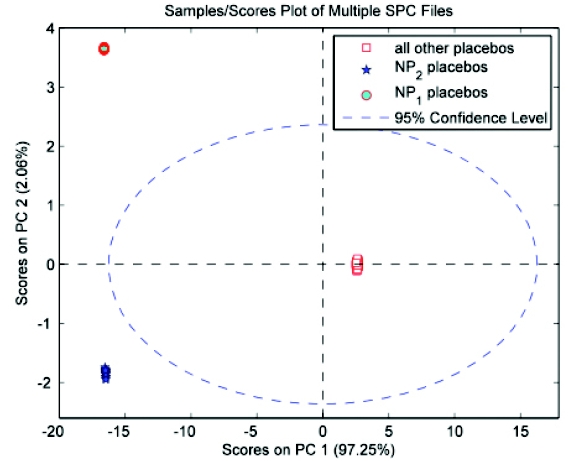 Figure 11 - Scores for PC 2 vs. PC 1 from PCA model of placebos with the addition of GLSW preprocessing (α = 0.1)
Figure 11 - Scores for PC 2 vs. PC 1 from PCA model of placebos with the addition of GLSW preprocessing (α = 0.1)- Identify classes – or even a single class – at each stage that can be readily separated from the others.
- Make conservative use of GLSW to down weight variables contributing to within-class variability.
- Once a model has been built that successfully discriminates one or more classes from the remaining samples, those classes no longer need to be included for analysis at the subsequent levels. This reduces the load during those steps by having to account for less sources of variability.
The first step in constructing the hierarchical model is to classify each sample according to one of the following groups:
- capsule placebo from one of the original products
- tablet placebo from one of the original products
- placebo from either NP1 or NP2
- active
Next, the following PLSDA models were built (in the following order)2 :
Capsule placebos
Building upon the results in Figure 11, complete separation of the capsule placebos from all other classes – including the new products – proved to be successful with the addition of GLSW preprocessing (Figure 12)
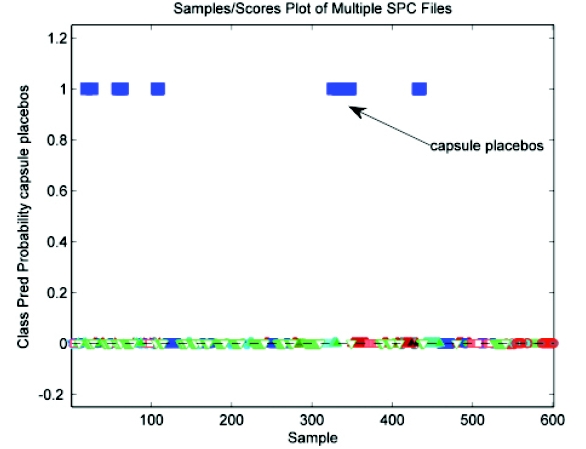 Figure 12 - Class predicted probability - PLSDA model with capsule placebos vs. all other classes
Figure 12 - Class predicted probability - PLSDA model with capsule placebos vs. all other classesClasses for modeling – capsule placebos (I); all other classes (II)
Classes excluded: none
Tablet placebos
Excellent separation was achieved in classifying tablet placebos from the remaining samples (actives and placebos of NP1 and NP2) as shown in Figure 13.
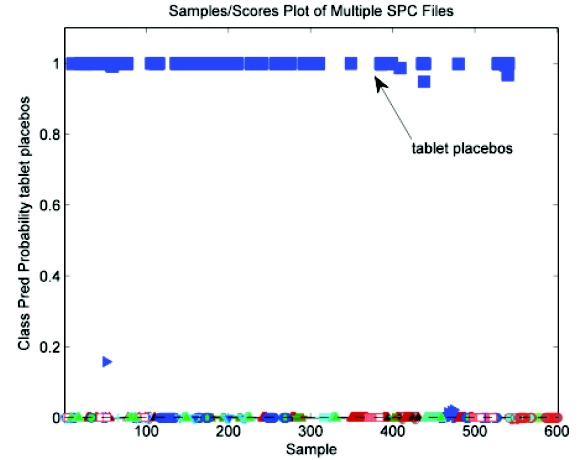 Figure 13 - Class predicted probability - PLSDA model with tablet placebos vs. actives and placebos from new products
Figure 13 - Class predicted probability - PLSDA model with tablet placebos vs. actives and placebos from new productsClasses for modeling – tablet placebos (I); actives, NP1 placebos, and NP2 placebos (II)
Classes excluded: capsule placebos
Placebos for NP1 and NP2
Having accounted for tablet and capsule placebos from the original products, the next step was to build a model that discriminated placebos from the new products against all actives. This model was somewhat less deterministic than the previous two, yet still useful. As can be seen in Figure 14, the discrimination between NP1/NP2 placebos and all of the actives is successful with the exception of the NP2 active group. Hence, if an unknown sample is predicted as belonging to the group from this model, there is still some uncertainty. On the other hand, there is a very high degree of certainty in a sample being predicted as an active at this stage.
 Figure 14 - Class predicted probability – PLSDA model with NP1/NP2 placebos and all actives
Figure 14 - Class predicted probability – PLSDA model with NP1/NP2 placebos and all activesClasses for modeling – NP1 and NP2 placebos (I); actives(II)
Classes excluded: tablet and capsule placebos
Last tier model – NP1/NP2 placebos vs. NP2 active
A sample that makes it to the third tier model that is designated as a placebo could actually be an active of NP2. Figure 15 provides some insight as to why this might be the case. The only significant difference between the NP1/NP2 placebo and NP2 active classes are very minor features at 1506 cm-1 and 1615 cm-1. Adding a fourth tier model that differentiates between NP1/NP2 placebos and NP2 actives is quite definitive (Figure 16); hence, an output of “placebo” for the third tier model should more accurately be considered as “placebo – maybe” with the fourth tier model providing a much more conclusive designation.
 Figure 15 - Comparison of mean spectra of NP2 active class and the NP1/NP2 placebo class
Figure 15 - Comparison of mean spectra of NP2 active class and the NP1/NP2 placebo class Figure 16 - Class predicted probabilities - PLSDA model with NP1/NP2 placebos and NP2 actives
Figure 16 - Class predicted probabilities - PLSDA model with NP1/NP2 placebos and NP2 activesClasses for modeling – NP1 and NP2 placebos (I); NP2 active (II)
Classes excluded: tablet and capsule placebos, all actives except NP2
The final hierarchical approach is summarized in the flowchart in Figure 17.
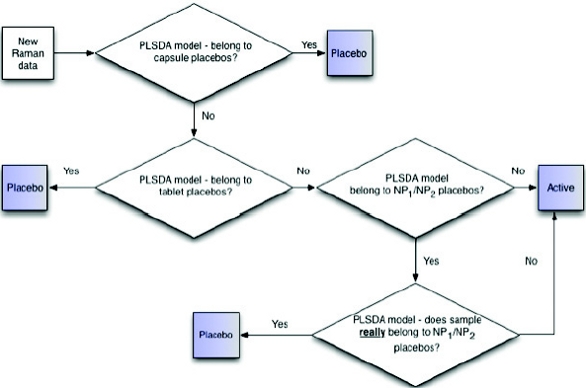 Figure 17 - Flowchart for final hierarchical model to classify placebos and actives
Figure 17 - Flowchart for final hierarchical model to classify placebos and activesThe established hierarchical model is ideal for classifying 14 different drug products and their placebos; however, testing new products would require further evaluation to determine if the products are properly classified. In one instance, new placebo products sharing a similar unit formula as those included in the model should be accurately predicted and may not require their addition to the model. Likewise, the active products should be evaluated for significant API spectral contributions to determine if they will provide an appropriate response for the model to classify as an active. In the final scenario, and similar to products NP1 and NP2 where the products and placebos share the same formulation, the model is recommended to be updated with the inclusion of the new products and their placebos which may require additional hierarchical steps for accurate classification. Simply put, there is no universal answer to the question as to whether the classification model in place needs to be updated with the introduction of new products to the slate; this needs to be addressed on a case-by-case basis.
Conclusion
Implementation of a chemometric Raman spectral library for positively identifying placebos offers several benefits. Unlike a product-specific negative HPLC identification test, a chemometric spectral library testing approach offers a specificity advantage by comparing sample results to several products at once. In addition, because transmission Raman testing is executed on whole tablets and capsules, laboratory analysis time is reduced (e.g., shorter setup, less instrument run time, simpler documentation, etc.). In our example, the testing of the calibration, validation and test samples set (400+) required approximately three days. Once the model was completed as a hierarchical model, testing demonstrated 100% correct classification of placebos and actives which included 14 different products and their respective placebos (capsules and tablets) at multiple strengths where active strengths ranged from 0.5 mg (0.25% drug load) to 300 mg (30% drug load).
References
- H. Martens, M. Høy, B.M. Wise, R. Bro and P.B. Brockhoff , “Pre-whitening of data by covarianceweighted pre-processing,” J. Chemom., 17(3), 153-165, 2003. Shaver, J.M., Gallagher, N.B, Wise, B.M., “Soft vs OrthogonalizatSoft vs. Hard Orthogonalization Filters in Classification Modeling,” Federation of Analytical Chemistry and Spectroscopy Societies, Louisville, KY, October 18–22, 2009.
Michael Dotlich, M.Sc., is a Research Scientist in analytical research and development at Eli Lilly and Company. He works in the Lilly Research Laboratories validating methods of testing for clinical trial materials release. His active research is focused on the development of spectroscopic methods using different analytical techniques and chemometrics for identification and quantitation of raw materials and drug products. He earned his M.Sc. in applications of Raman spectroscopy from Marquette University, Milwaukee WI.
Dr. Bob Roginski is a Senior Applications Scientist with Eigenvector Research, Inc., where he provides consulting services, instruction, and software development in the area of chemometrics. Previously, Bob served in engineering roles specializing in process analytical technology at Eli Lilly & Co., Searle/Pharmacia/Pfizer, and Amoco Corporation. Bob received his Ph.D. in Chemical Engineering from the University of Illinois in 1987, and has collaborated on numerous peerreviewed publications and outside presentations. Bob has special interests in spectroscopy as applied to PAT and using chemometrics to determine the health of continuous processes.
Richard M. Kattner, M.Sc., is Associate Consultant Chemist in the analytical research and development at Eli Lilly and Company. His current work in the Lilly Research Laboratories deals with developing and validating methods for the testing and release of clinical trial materials. His current focus is the development and validation of spectroscopic methods using different analytical techniques and chemometrics for testing active/placebo tablet and solutions. He earned his M.Sc. in Chemistry focusing in Physical Organic Chemistry at the University of North Texas, Denton, TX.
Dr. Jeremy Shaver is currently the Chief of Technology Development at Eigenvector Research, Inc., which he joined in 2001. He received a BA in Chemistry from the College of Wooster in 1991 and a Ph.D. in Analytical Chemistry from Duke University in 1995.
1 IFPAC 2013 Presentation – Enhanced Classification Using Fused Data, Michael Dotlich, Eli Lilly, Bob Roginski, Jeremy Shaver, Eigenvector Research
2 Data over the spectral range of 201.7-1701.6 cm-1 was used for each model with the following preprocessing steps: 2nd derivative (15 pt. window, 2nd order polynomial), SNV, GLS weighting (α = 0.1), and mean centering