Introduction
Low endotoxin recovery (LER) was first described publicly at the PDA Annual Meeting in Orlando Florida, in April, 20131 but the phenomenon has been observed by pharmaceutical scientists for years, mostly as classical “inhibition” during sample qualification testing.
The U. S. Food and Drug Administration (FDA) Guidance for Industry Pyrogen and Endotoxin Testing: Questions and Answers was published in 2012.2 The third question posed by the FDA in the guidance pertains to establishing the stability of endotoxins in products. The FDA’s response to this question was: “The ability to detect endotoxins can be affected by storage and handling. Firms should establish procedures for storing and handling (which includes product mixing) samples for bacterial endotoxins analysis using the laboratory data that demonstrates the stability of assayable endotoxins content. Protocols should consider the source of the endotoxin used in the study, bearing in mind that purified endotoxins might react differently from native sources of endotoxins.”
Native Endotoxins and Purified Lipopolysaccharide
Purified Lipopolysaccharide and native endotoxins are not the same material. A brief description of the terms lipopolysaccharide, native endotoxins, and low endotoxin recovery, also known as endotoxin masking, are as follows:
Lipopolysaccharide (LPS) is the biologically active component of the endotoxin molecule. Well known examples of a highly purified LPS derived from Escherichia coli O113:H10 are the primary standard (Reference Standard Endotoxin or RSE) obtained from the USP and the secondary standard (Control Standard Endotoxin or CSE) obtained from lysate suppliers for the compendial Bacterial Endotoxins Test (BET) used to standardize Limulus amoebocyte lysate (LAL) reagents and perform compendial suitability testing.
Purified LPS is not true endotoxin but a fraction of the cell wall fragment of Gram-negative bacteria. In contrast, native endotoxins are components of the outer cell walls of Gram-negative bacteria. They exist naturally as vesicles, which contain LPS, outer membrane surface proteins, lipoproteins, and phospholipids. Native endotoxin exists as bacterial cell wall fragments and is known as an extremely stable high molecular weight molecule resistant to heat and chemical destruction.
LER is also referred to as the phenomenon of endotoxin masking. LER is the inability to recover a spiked amount of LPS in a study designed to demonstrate the stability of assayable endotoxins. The masking occurs when LPS is added directly to the sample prior to preparing sample matrix dilutions for testing. LPS disappears, and is not recoverable when assayed.
Industry studies are demonstrating that native endotoxin does not exhibit an LER effect. In contrast, studies are demonstrating that LPS does exhibit an LER effect, especially when placed directly into biological monoclonal matrices containing cation-chelating citrate and phosphate buffer systems with polysorbate surfactants. Therefore this phenomenon, as pointed out by lysate expert Dr. James Cooper, is really low lipopolysaccharide recovery (LLR) rather than LER.3 The criterion for determining if LLR is demonstrated for the experiments shown in this article was <50% recovery of the starting concentration of analyte.
Industry Response
There was some confusion in the industry about the FDA expectations for addressing the stability of endotoxins, especially in biological products. Initial interpretations were that if a product was found to have no “assayable endotoxin,” the testing lab did not have a compelling reason to demonstrate “stability of assayable endotoxins.” After all, there was no analyte (endotoxin) present at zero time so there was no stability issue. Because purposely contaminating a product with a purified LPS such as CSE or RSE did not appear to be a workable experimental design, many pharmaceutical companies moved forward with creating a standardized natural endotoxin to spike product. An FDA reviewing microbiologist was asked a clarifying question in an open forum in Philadelphia at the Bacterial Endotoxins Summit in May, 2014. Based on his answer, we were lead to believe that the original intent of the FDA response for the third question was not to artificially contaminant samples with endotoxin to demonstrate stability of assayable endotoxins. In retrospect, it does not appear prudent to execute spiking experiments with LPS, knowing the labeled instructions for storing reconstituted CSE and RSE. In its reconstituted form, the RSE recommended hold time at 2-8°C is 14 days. The CSE, in its reconstituted form, has a longer labeled shelf life (recommended from most vendors) of 30 days at 2-8°C. Most microbiologists are conservative and discard their diluted RSE or CSE standards at the end of each day. Furthermore, the knowledge that RSE and CSE disappeared over time in chelating buffers was not a surprise to most microbiologists qualifying endotoxin assays as required by USP <85> Bacterial Endotoxins Tests. Inhibition and enhancement testing already demonstrated that the many product matrices could not be tested undiluted.
Demonstrating stability of the RSE and CSE spiked into product is inappropriate for companies conducting LER experiments. Current evidence indicates that the loss of LPS activity in certain biological matrices or platform buffering systems used for monoclonal antibodies containing polysorbate and a chelating buffer occurs rapidly. Most tellingly, the product protein does not have to be present for the measurable loss of CSE or RSE. In practice, LER occurs immediately during the preparation of the bulk biological product, ie, upon addition of the polysorbate to a citrate or phosphate buffer and the purified protein. Possible corrective actions such as no delay in laboratory testing or adjusting endotoxin limits downwards to accommodate losses in recovery are not viable solutions. In many cases, “endotoxin masking” is close to a 100% loss of spiked LPS.
Do these findings indicate that our use of purified LPS as CSE and RSE are unsuitable for their intended use as standards? Absolutely not. An earlier batch of RSE was standardized against the rabbit pyrogen test in the early 1970’s.4 The CSE distributed by lysate manufacturers is qualified for the BET as secondary standards which are calibrated against the primary RSE. The RSE is in limited supply and is too valuable a resource to be used in routine endotoxin testing. The purpose of standards is to quantify the amount of endotoxin in a product, to evaluate the test values against the established acceptance criteria, and to perform suitability testing. The BET and these standards have served the industry extremely well over the years as pyrogenic reactions in patients are extremely rare. There is no evidence of a patient safety concern associated with the current compendial BET. It is well documented that the BET is a more sensitive and robust predictor of pyrogenicity than the rabbit pyrogen test.4,5 During the development and standardization of the compendial BET, there were no false negative tests. In every case, the BET was able to detect and measure endotoxin pyrogens at levels that rabbits did not detect.
The suitability test conducted during sample qualification of the BET is executed by “spiking” a known amount of CSE or RSE into the suitably diluted sample matrix to demonstrate that the endotoxin detection mechanism (gel clot, turbidity formation, or color formation) was not inhibited or enhanced. For some products, dilution is unnecessary. For most products, however, dilution is necessary to overcome test interferences. Validity for gel clot assays is gelation in a 2λ positive product control. Validity for quantitative assays is 50-200% recovery of the positive product control. The suitability test in not intended to qualify endotoxin recovery directly from the sample but after dilution into the non-interfering and assayable range. Most compendial biological test qualifications verify sample suitability in this manner. For instance, when conducting a suitability test for the compendial sterility test, one typically adds the organisms to the final rinse and not directly to the test sample.
It should be emphasized that LLR occurs when RSE or CSE is placed directly into the test sample matrix without dilution. The details for the mechanism of the “masking effect” are not well described. At this time, the “masking” in biological matrices has been limited to citrate and phosphate buffers with chelating properties which contain polysorbate surfactants. The products evaluated thus far because of their prominence in the drug pipeline have been mostly monoclonal antibody formulations. One hypothesis for LLR is that purified LPS may no longer be recognizable by the LPS binding receptor of Factor C. Therefore, no activated Factor C can trigger the enzymatic cascade that leads to gel clotting.7 Another probable explanation with some supporting data has been proposed by Petsch et.al.3, 6, 8 The proposed mechanism is that the chelating buffers remove the divalent cations, especially Mg+2, from the LPS molecule causing destabilization of micelles resulting in disaggregation to a less LAL-reactive, monomeric, and unrecognizable form. A pictorial description or cartoon of this event can be viewed in the presentation from the Berlin Endotoxin Workshop.9 Many product formulations exist which may demonstrate an inability of LPS recovery when placed directly into product matrices as an artificial contaminating analyte. Many of the interfering matrices are known to have low or high pH, chelating buffers, or other interfering factors that can render an LPS molecule unreactive in the BET.
Using purified LPS to demonstrate “stability of assayable endotoxin” is an unadvisable strategy. The demonstration should be conducted with native endotoxin. There are some products including vaccines that have endogenous endotoxins either as part of their active ingredient or as contaminants from raw materials or processes. These products fall mostly into the biological category. It may be valuable to demonstrate the “stability of assayable endotoxins” to assure that the finished products meet the same endotoxin limits at the end of shelf life as products do for the release BET. However, this author questions the value of artificially contaminating a non-endotoxin containing product to demonstrate “stability of assayable endotoxins” when none exist.
Experimental Evidence for LER
The following experiments were conducted to demonstrate that LER does not exist whereas LLR does exist in the presence of chelating buffers containing polysorbates. The experiments used a kinetic turbidimetric test method. The CSE (LPS) was freshly prepared at Time=0. The native endotoxin (derived from the Gram-negative bacterium EnterobacterMs. Platco has been with Merck & Co. for 29 years. She is currently a Principal Scientist performing endotoxin and microbiological testing for all pharmaceutical and vaccine/ protein products. She has been an active participant in the LAL User’s Group and is a member of the PDA Microbiology Annual Meeting planning committee moderating sessions dealing with endotoxin issues.
cloacae, at approximately1000 EU/mL) was supplied by the lysate supplier. The standard curve values used for measurement were 5.0, 2.0, 0.5, 0.125 and 0.05 EU/mL (lambda). All samples and test dilutions were prepared in pyrogen-free glass test tubes. The samples were held at 2-8°C between test intervals unless otherwise noted. All results are reported as EU/mL (IU/mL).
The initial experiments involved spiking CSE (LPS) and native endotoxin into a range of monoclonal antibody buffer matrices and observing the change in LPS and endotoxin concentrations measured by the BET over 15 days.
Experiment One
The first experiment was conducted with a 10-mM sodium phosphate buffer solution and adding polysorbate 80 at 0.01% and 0.02%. A pyrogen-free water control (PFW) was also spiked and tested. To confirm suitability testing for this buffer, 1:10 dilutions of the solutions were prepared in PFW as this dilution had previously been the valid routine test dilution demonstrated. The undiluted solutions (10 mL for CSE [LPS] and 5 mL for endotoxin) were spiked with 200 mcL of CSE (LPS) and 100 mcL of endotoxin at approximately 1000 EU/mL. The expected concentrations for both CSE (LPS) and endotoxin were approximately 20 EU/mL. The results indicate that the sodium phosphate buffers containing polysorbate 80 did exhibit a loss of CSE (LPS) reactivity overtime (Figure 1). The buffers that were spiked with the native endotoxin did not demonstrate a reduction in reactivity over time (Figure 2).
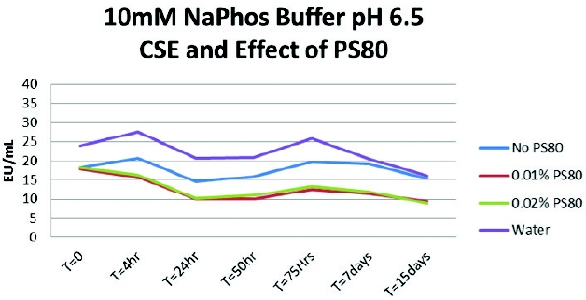 Figure 1 - The Effect of Surfactant Concentration on LPS Spike Recovery
Figure 1 - The Effect of Surfactant Concentration on LPS Spike Recovery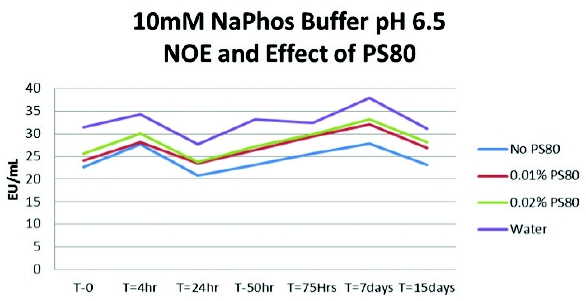 Figure 2 - Effect of Surfactant Concentration on Native Endotoxin Spike Recovery
Figure 2 - Effect of Surfactant Concentration on Native Endotoxin Spike RecoveryExperiment Two
A second experiment was conducted with two similar monoclonal antibody (mAb) formulations at 25 mg/mL protein in 20-mM citrate buffer. The buffer also contained 150 mM saline and 0.025% (v/v) polysorbate 80. To confirm suitability testing for these two mAb, 1:10 dilutions of the mAb were prepared in PFW as this dilution had previously been the valid routine test dilution. The undilute mAb solutions (10 mL for CSE [LPS] and 10 mL for endotoxin) were spiked with 75 mcL of CSE (LPS) and endotoxin at approximately 1000 EU/mL. The calculated concentrations for CSE (LPS) and endotoxin were approximately 7.5 EU/mL. The results demonstrate that CSE (LPS) is highly affected by the citrate buffer and polysorbate 80 while the true endotoxin recovery was unaffected (Figure 3). Additional studies (not shown) also demonstrate that the “masking” effect with CSE (LPS) recovery begins almost immediately after addition to the undiluted product matrix. More studies were performed in an effort to try to reverse the “masking” using an endotoxin dispersing reagent and replacement of divalent cations with little success.
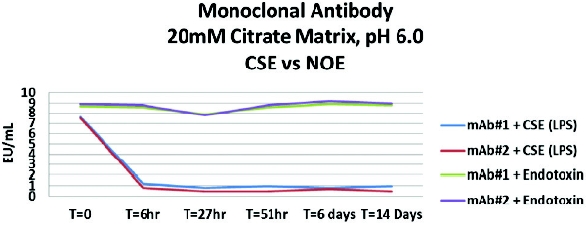 Figure 3 - Effect of Citrate Buffer Matrix on LPS and Native Endotoxin Spike Recovery
Figure 3 - Effect of Citrate Buffer Matrix on LPS and Native Endotoxin Spike RecoveryThe loss of CSE (LPS) reactivity in a mAb occurs within 6 hours in the presence of citrate and phosphate buffers containing polysorbate. The loss of reactivity appears to be temperature dependent when the experiment was repeated during a 4-hour period at 2-8°C, room temperature and 30- 35°C. The data indicate that the masking phenomenon occurs faster at higher temperatures (Figure 4). A revealing additional experiment was conducted whereas endotoxin-dispersing reagent was added prior to adding the CSE (LPS) contaminant to the undilute matrix. The results indicate that under these conditions, the masking effect did not occur. However, adding endotoxin dispersing reagent to the already masked CSE (LPS) could not reverse the LLR effect, even if stored at 30-35°C overnight (data not shown).
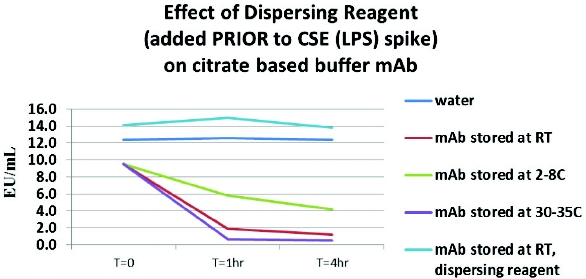 Figure 4 - Effect of Temperature and Dispersing Agent on LPS Spike Recovery
Figure 4 - Effect of Temperature and Dispersing Agent on LPS Spike RecoveryOther Platform Buffering Systems
Another common buffer for biological matrices is histidine buffer, which does not have the chelating effect of the citrate and phosphate buffers. An experiment was conducted on a mAb in a histidine buffer, which is lyophilized and reconstituted for testing. The reconstituted sample matrix was 25 mg/mL protein in 10-mM histidine buffer containing 7% sucrose and 0.02% polysorbate 80. The undiluted monoclonal antibody solutions (10 mL for CSE [LPS] and 10 mL for endotoxin) were spiked with 200 mcL and 75mcL of CSE (LPS) and endotoxin at approximately 1000 EU/mL. The expected concentrations for CSE (LPS) and endotoxin were approximately 20 EU/mL and 7.5 EU/mL. The 20-EU/mL spiked samples were diluted 1:50 for testing (routine test dilution) and the 7.5 EU/mL spiked samples were diluted 1:10 for testing (maximum valid concentration of product). PFW controls were also spiked (data not shown).
This matrix demonstrated that neither CSE (LPS) nor native endotoxins are affected and recoveries were all stable throughout the test time period of 15 days (Figure 5).
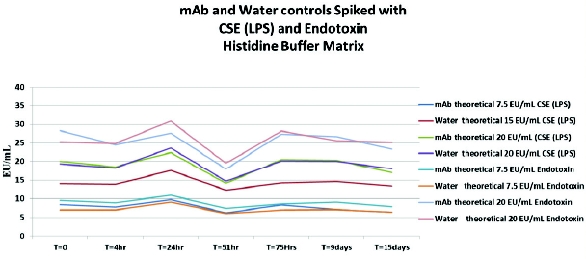 Figure 5 - Effect of Histidine Buffer Matrix on LPS and Native Endotoxin Spike Recovery
Figure 5 - Effect of Histidine Buffer Matrix on LPS and Native Endotoxin Spike RecoverySummary and Conclusions
The data indicate that purified LPS and native endotoxin react differently in spike and recovery experiments in different biologic formulations.
Native Endotoxin and not purified LPS is the reliable a predictor of stability of the endotoxin, especially in the presence of chelating citrate and phosphate buffers containing polysorbate surfactants. The exact mechanism of the LPS “masking” effect is unknown, but as reported by other investigators, is the likely result of a destabilization of micelles. Laboratory studies reported in this article clearly demonstrate that the low endotoxin recovery phenomenon is an artifact of study design and sample matrix. Low endotoxin recovery (LER) is not a real phenomenon as native endotoxin is not masked. The author supports the position proposed by J. Cooper that this artifact should be called LLR. The compendial BET continues to be a highly sensitive, accurate and robust assay for measuring bacterial endotoxins. Despite the recent highlighting of the LLR phenomenon, injectable pharmaceutical and biological products remain safe.
References
- Chen, J. “Low Endotoxin Recovery in Common Biologics Products.” Presented at the 2013 PDA Annual Meeting. Orlando, FL, April 2013.
- Guidance for Industry Pyrogen and Endotoxins Testing: Questions and Answers, U.S. Food and Drug Administration. June 2012.
- Cooper J. “How Can Water Interfere with the BET Test.” Presented at PMF Bacterial Endotoxin Summit. Philadelphia, PA, USA. May 15-16, 2014.
- Williams KL. Endotoxins as a Standard. In: Endotoxins: Pyrogens, LAL Testing and Depyrogenation. Drugs and the Pharmaceutical Sciences, Volume 111. 2nd ed. Marcel Dekker Inc. New York, USA. 2001: 136-164.
- Mascoli C, Weary M. Applications and advantages of the Limulus Amebocyte (LAL) Pyrogen test for parenteral injectable products. In: Biomedical Applications of the Horseshoe Crab (Limulidae). Liss AR. 1979:381-402.
- Dubczak J. “LAL: A Comparative.” Presented at PMF Bacterial Endotoxin Summit. Philadelphia, PA, USA. May 15-16, 2014.
- Chen J, Williams K. Follow-Up on Low Endotoxin Recovery in Biologicals. PDA Letter. October 2013; XLIX (9):14, 16.
- Petsch D, Anspach FB. Endotoxin Removal from Protein Solutions. Journal of Biotechnology. 1991;76 (2000):97-119.
- Reich J. “Reliability of Endotoxin Detection Mechanical principles of Endotoxin masking and Strategies for Demasking.” Presented at the 2014 Preconference Workshop on bacterial Endotoxin Testing. Berlin, Germany, February 2014.
Ms. Platco has been with Merck & Co. for 29 years. She is currently a Principal Scientist performing endotoxin and microbiological testing for all pharmaceutical and vaccine/ protein products. She has been an active participant in the LAL User’s Group and is a member of the PDA Microbiology Annual Meeting planning committee moderating sessions dealing with endotoxin issues.