Introduction
Forced degradation studies (FDS) are essential in the development of stability indicating methods to gain understanding of the intrinsic stability characteristics of a Drug Substance (DS). Performance of stress studies under various conditions allows prediction of the possible degradants that may be obtained during registrational long term stability studies (LTSS). These degradants are used to develop and validate the selectivity required for registrational DS and drug product (DP) analytical methods. New molecular entity registrational filings require discussion of forced degradation data in stability indicating methods (SIM) validation and degradation mechanistic stability sections along with any relevant controls in both synthesis and the formulation sections of the Health Authority dossier submission.
Guidance on forced degradation used by industry comes via formal documents from ICH1, FDA2, and publications/reviews on the topic3-5. Recently, ANVISA (the Brazilian National Health Surveillance Agency) published the RDC53/2015 regulation outlining specific requirements for product registration and post-approval change submissions with regard to reporting, identification and qualification of degradation products6 and the associated guide7 with expected information to be included in the report. These are generally aligned with industry practices, however, the scope and depth of such requirements are expanded beyond the ICH guidelines detailing specific requirements and recommendations for FDS. The extension of FDS requirements necessitated a review and update of current accepted practices, and assessment of its impact to previously filed and registered products to ensure compliance.
ANVISA primarily focused on registrational filing requirements for FDS, specifically to obtain: 1) Identification of functional groups susceptible to major degradation pathways and subsequent mechanistic understanding, 2) Assessment of specific FD experimental conditions and extent of degradation (Articles 4, 5 and 6 of the RDC53/2015), 3) The requirement for specific information to be captured within a formal report, 4) Consideration of degradants resulting from drug product manufacture and storage, 5) Safety qualification of degradation products and 6) Compliance with the new regulation for approved products and post-approval change submissions.
The forced degradation requirements are discussed herein in relation to study design, reports and interpretation of such requirements. Expectations pertaining mostly to performing and reporting the forced degradation study from the viewpoint of a drug manufacturer are detailed.
Chronology of Degradation Studies
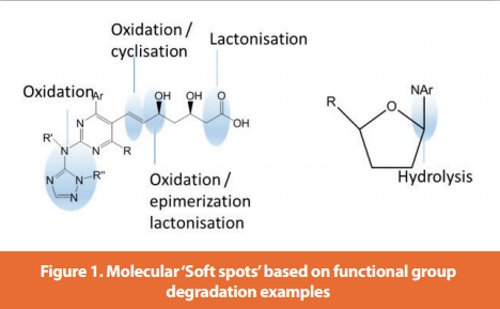
In pre-development, a limited scope (general) FDS is typically performed for discovery candidate nominations. Additional and more focused molecular stability studies for discovery compounds transitioning into development may uncover known chemical conditions of instability. Degradation can be further understood with an evaluation of the chemical structure to determine likely high risk functional groups or ‘soft spots’ based on common known degradation pathways8,9 (e.g., carboxylic acid or amide groups) as shown in Figure 1. Molecular degradation predictions may also be evaluated using specialized software10. Furthermore, DS/excipient compatibility studies are used when scouting possible early formulations to control/ minimize the risk of degradation through formulation design. Additional controls may be put in place based on this knowledge, e.g., with use of appropriate packaging technologies such as the use of desiccants or antioxidants. Further knowledge may be obtained when performing stability assessment of DS, e.g. solid state form studies. Any significant degradation should be further investigated to determine degradant structure and mechanism of formation. Overall this foundation of knowledge provides a good starting point for a structural physico-chemical evaluation and, subsequently, a more tailored forced degradation protocol design for later stages of development. The forced degradation extent as a function of the pharmaceutical development timeline is illustrated in Figure 2.
Stability Indicating Method Development
The development of stability indicating methods (SIM) relies on the execution of appropriate forced degradation studies to obtain the relevant degradants required to establish and validate selective chromatography for long term registration stability studies and release testing5. Across the pharmaceutical industry internal guidelines tend to be used to ensure studies are performed consistently11 and capture specific product historical knowledge. It is essential that these studies are performed on both drug substance and drug product made from the processes intended for commercialization. The appropriate balance of forming primary degradants versus the unrealistic and unnecessary formation of secondary and tertiary degradants (degradants of degradants) is considered in the extent of degradation targeted4 (typically 5-20%). A well thought-out study design including past knowledge will focus the study protocol and minimize the necessity of repeated work. Decision trees and kinetic studies can be used to assist in the design of a simple study, appropriate for the intended purpose of generating relevant degradants. There is a need to balance the method needs vs. experimental constraints and typically a kinetic study will help in covering the level of degradation required along with deciphering mechanisms for primary vs. secondary degradation. With this information as a starting point (including earlier phase I-II pharmaceutical development work) coupled with performing appropriate characterization, a mechanistic understanding of the major degradation pathways may be proposed. This collective information is typically outlined in the stability section of the registrational submissions to help rationalize the stability trends observed.
ANVISA FDS Requirements
ANVISA RDC 53/2015 has specific requirements for FDS at its core. These requirements are described in articles as summarized in Table 1. Article 4 - what material should be studied, Article 5 - what condition must be used and Article 6 - what results are acceptable and how their use in SIM development. An example of internal guidance for FDS is shown in Table 2. This approach has been aligned with the ANVISA RDC 53/2015 (specifically articles 4, 5 and 6), and it contains details to assist in the study design that can be used to help develop an understanding of molecular stability or instability. Additionally, a stepwise guidance, as illustrated with the acid/base decision three example (Figure 3), compliments the guidance and brings about consistent work practices, helping the user reach the desired endpoint of the study and mitigate problems such as side reactions (potentially distracting information). Consistent and value-added studies assist in SIM development, as well as provide the foundation of mechanistic understanding.
There are several specific aspects of FDS that require more detailed explanation that have been discussed in the literature and in RDC- 53/2015. These are discussed in the following sections.
Placebo Use and Control Materials
The FDS should be applied to placebo, formulation and isolated DS(s) for all conditions, and a control (reference standard) should be included in any analysis. Fixed-dose combinations should be tested as a combination of DSs and in the formulation for all conditions. However these studies may not be required concurrently if previous study information can be applied e.g., if previously performed using the appropriate DS crystalline form that is representative of the final process.
Solvent and Environment
Organic solvents may be needed to aid solubility of DS under one or more FD conditions to obtain the required exposure. However potential side reactions due to the organic additive must be taken into consideration12. It is recommended to consider appropriate solvents e.g. water soluble radical initiator AAPH (2,2’-azobis (2-aminopropane) dihydrochloride) vs. AIBN (2,2’-azobis(2-methylpropionitrile)) that requires organic solvent. Additionally, any precipitates will need to be dissolved during sample preparation to ensure the detection of poorly soluble degradants and to maintain mass balance.
Acid and Base Studies on Drug Product
Drug product acid/base stress studies are required, based on the following ANVISA rationale6 “...even knowing that the active pharmaceutical ingredient of a solid dosage form will not be exposed to any acids during its storage, the degradation condition in acidic environment may accelerate reactions with the excipients that would only be noticed after long periods of time under normal conditions”. In the case where hydrolytic degradation is observed from the DS stressed studies, acid/base stress testing should also be performed for the drug product to determine if any new degradation products are formed due to interaction with the formulation excipient matrix.
Metals
Metals, such as Fe (III) and CU (II)13, at a minimum should be used to assess the DS susceptibility to metal catalyzed oxidation and if any degradation is observed, the study should be repeated for the drug product. If alternate scientific data demonstrating stability, e.g., results from drug-excipient compatibility studies are already available, it could be used to justify why the studies are not performed on DP.
Amount of Degradation Required
Typically 10-15% degradation is desired as a FDS endpoint for the different FD conditions. Clearly not all compounds / formulations will degrade by 10% under even extreme conditions, however a sufficient attempt must be taken to promote degradation. Any stability observed from the studies must be scientifically explained and justified in case of low degradation levels (i.e. < 10%). These types of criteria can be used within internal guidance documents where extreme conditions are defined.
Study Duration
The extent to which FD studies can be accelerated has been discussed in the literature3 , and in particular, the appropriate duration of the studies. Most justification is based on the rule of thumb where a 10 °C increase in temperature is conservatively considered to give a doubling of the degradation rate based on the Arrhenius equation (assuming a first order reaction). This is considered to be a reasonable approximation up to 70 °C14. At temperatures greater than 80 °C, the increased activation energies could promote secondary or alternate unrealistic pathways, which become problematic and may complicate the degradation pathways and profiles too much. Based on the authors’ experience, it is more informative to extend studies to one month rather than increase temperatures to 80 °C or above.
Data Assessment
In terms of method validation, forced degradation studies provide confirmation on specificity of the developed SIM. The obtained data are assessed to ensure adequate separation of degradation impurities from the active (peak resolution), no co-elution of degradation impurities with the active (peak purity) and capability to detect all potential degradation impurities (mass balance). The FDS information helps support formulation, packaging and storage condition selection. When studying fixed-dose combinations, it is important to compare results from individual active components along with the combination for both DSs and formulations.
Mass Balance
Assay of the DS and formulation samples should be performed. The assay, corrected for the degradants and impurities, should be calculated. Investigation of mass imbalance, e.g., >5%, depending on number of impurities observed (only a few large peaks may be simpler to explain vs. many small peaks,) should be conducted and justified especially when large response factor (RF) diff erences are expected (e.g., a non-UV absorbing portion of the molecule is a degradant). Obtaining mass balance may be challenging depending on RF differences or insolubility or volatility of degradants or process residual solvents, however a reasonable attempt is expected and any conclusions must be scientifically justified.
Characterization of Degradants
Typically, full structural elucidation is only performed for significant degradants. This will help confirm the degradation mechanism and understand the key degradation pathways. Such identified degradants may require initial in-silico assessment for toxicity and subsequent qualification, if observed at high levels in accordance with ANVISA RDC-53/2015 and ICH Q3B(R2). Additionally, these are the most likely degradants to be formed (if at all) in the long term stability study, and are, therefore, necessary for the selectivity validation element of the stability indicating analytical HPLC method. Small amounts of the material will need to be synthesized or isolated, with their purity obtained for RF determination and accuracy studies.
Study Reports
Although ICH and FDA do not have any specific guidance on registrational filing reports, ANVISA had issued what may be considered prescriptive / detailed recommendations. These include:
- The introduction should contain discussion on active functional groups susceptible to degradation.
- The forced degradation experimental section should focus on the conditions used for the satisfactory levels of degradation or the scientific justification of why degradation did not take place.
- Overlay of chromatograms from each degradation condition at 125% and 5% scale based on main peak height. These should include the blank, control (initial or unstressed study material) and placebo to compare degradation profiles. Specific data with chromatograms including percent degradation, resolution, column efficiency, peak purity and tailing factor for each peak.
- Additionally a discussion on mass balance, study results and degradation mechanisms should be included.
- The conclusion should contain a qualitative degradation profile, production conditions vs. degradation profile and storage implications.
Impact on Marketed Products
RDC No. 53/2015 also applies to products already registered in Brazil, where the market license holders are responsible for ensuring compliance with the provisions set forth in this regulation by the effective dates according to drug classifications outlined in paragraphs I, II and III of Article 14. FDS must therefore meet Article 4 – 6 requirements. For mature brand products, FDS would typically have followed regulatory guidance in effect at the time of filing, with less details and requirements than those provided in the RDC No. 53/2015. Consequently certain data, such as mass balance, may not be readily available and would require extensive work to obtain, if necessary to do so. However, the efficacy and safety of mature brands have been well established. Extensive knowledge, stability data and safety experience have been obtained through drug product formulation development, and shelf-life (pre and post marketing) stability studies for both drug substance and drug product. This body of data, along with appropriate scientific rationale, may be used to justify why certain FDS requirements could be waived.
The following steps for a consistent assessment of marketed products are recommended: 1) Review of available degradation profile data vs. Article 4 – 6 requirements. 2) Perform an impact assessment on missing elements in terms of safety, therapeutic efficacy or degradation control risks. 3) If considered low or no risk, provide scientific justification in the petition document (Table 3 example). If missing element is assessed as high risk, appropriate actions which may include, as required, further FDS, method optimization and revalidation may be considered.
Generally, an assessment of previously performed studies for approved products should result in low or no risk, since established safety records in conjunction with proven stability have been demonstrated throughout the shelf-life (an example for a product is shown in Table 3). Examples of scientific rationale resulting in low or no safety, inefficacy or degradation control risk assessment is provided in Table 4.
Summary
The ANVISA forced degradation study requirements will lead to an expansion of FDS beyond the work already performed during the various stages of development. The approach described will require a collaborative effort from development partners to achieve meaningful FDS design. Additionally, application of the findings will generate value by mitigating risks through systematically building on the molecule and formulated product knowledge throughout development.
The detailed structure and justification found in both the existing regulations and the new guides from ANVISA justify the requirement for a single report covering suitability assessment of the proposed stability indicating method, molecular structure review and its connection to the degradation profile, FDS, interpretation of data, and application of informed decisions / controls leading to the final formulation design, packaging selection and recommendation on storage condition. The report should include conclusions on the degradation profile with mechanisms and a safety profile assessment, and include assurances on degradation mitigation through production conditions and formulation storage controls.
Acknowledgments
The authors would like to thank the following R&D, Global Quality and Regulatory CMC colleagues for helping compile data and useful discussions.
Li Li, Nalini Anand, David Lloyd, Abdelhady Elkhouga, Catherine Leach, Shan Xiao, Xiao Cui, Yande Huang, Joan Ruan, Flavio Brino, Natania Sekine, Elizabeth Oliveira, Joel Young, Mike Ribick, Ginna Mejia and Tania Lopes da Cunha
Disclaimer - These are the opinions of the authors based on the interpretation of the ANIVISA guidance.
References
- ICH, Q1A (R2), Stability Testing of New Drug Substances and Products (Nov 2003); ICH Q1B Stability Testing: Photostability Testing of New Drug Substances and Products (Nov 1996); ICH Q2B Validation of Analytical Procedures: Methodology (Nov 1996)
- FDA 21 Code of Federal Regulations, Part 211, cGMP in Manufacturing, Processing, Packaging, or Holding of Drugs and Finished Pharmaceuticals: 21 CFR 211.165(e): Testing and release for distribution; 21 CFR 211.166(a)(3): Stability testing; FDA Guidance for Industry, Analytical Procedures and Methods Validation for Drugs and Biologics, July 2015
- Pharmaceutical Research and Manufacturers of America Analytical Research and Development Steering Committee workshop, Reynolds, D.W., Facchine, K.L., Mullaney, J.F.,Alsante, K.M., Hatajik, T.D., Motto, M.G. “Available Guidance and Best Practices for Conducting Forced Degradation Studies,” Pharm. Technol. 2002, 26 (2), 48–56.
- M Blessy, M., Patel, R. D., Prajapati, P. N., Agrawal Y. K., Development of forced degradation and stability indicating studies of drugs. Review Paper. Journal of Pharma Analysis; 2014, 4(3), 159-165.
- Aubry, A.F., Tattersall, P., Ruan, J., Development of Stability Indicating Methods, Kim Huynh-Ba (Ed.) Handbook of Stability Testing in Pharmaceutical Development: Regulations, Methodologies, and Best Practices, Springer, NY., 2008, 139-151.
- ANVISA Brazil RDC 53 (2015) Regulation on report, identification and qualification of degradation products
- ANVISA Brazil Guideline No. 04 (2015) Guideline for obtainment of the degradation profile, and identification and qualification of degradation products in drugs
- Alsante, K.M., Ando, A., Brown, R., Ensing, J., Hatajik, T.D., Kong, W., Tsuda, Y., The role of degradant profiling in active pharmaceutical ingredients and drug products, Adv. Drug Del. Rev., 59, 29–37, (2007)
- S. Görög, Drug safety, drug quality, drug analysis, J. Pharm. Biomed. Anal., 2008, 48, 247–253.
- http://www.lhasalimited.org/products/zeneth.htm
- Alsante, K. M., Martin, L., Baertschi, S. W., A Stress Testing Benchmarking Study, Pharm. Technol., 2003, 27 (2), 60–73.
- Baertschi S.W., Jansen, P. J., Stress Testing: A predictive Tool, S.W. Baertschi (Ed.), Pharmaceutical Stress Testing-Predicting Drug Degradation, Taylor and Francis, NY., 2005, 24 & 461.
- Baertschi S.W., Jansen, P. J., Stress Testing: A predictive Tool, Baertschi S.W. (Ed.), Pharmaceutical Stress Testing-Predicting Drug Degradation, Taylor and Francis, NY., 2005, 27 & 221.
- Hotha, K. K., Reddy, S. P. K., Raju, V. K., Ravindranath, L. K., et al., Forced degradation studies: practical approach, Int. J. Pharm., 2013, 4 (5), 78-85.
About the Authors
Peter Tattersall, PhD is a Principal Scientist in Analytical and Bioanalytical Development department within Pharmaceutical Development at BristolMyers Squibb Company in New Jersey. He received his BSc. and Ph.D. from the University of Manchester, UK. He previously worked in Analytical Development at AstraZeneca, Wilmington. He joined Bristol-Myers Squibb in 2003. Where he is an analytical project team leader and supervises a small group of analytical chemists working on both drug substance and drug product development.
Suwimon Asawasiripong, PhD joined Bristol-Myers Squibb in 2014 as an Associate Director in the Global Analytical Technology (GAT) Department within Global Manufacturing & Supplies. She received her Ph.D. in analytical chemistry at Seton Hall University, NJ. Prior to joining BMS, she spent over 20 years in Analytical R&D positions of increasing responsibility at Purdue Pharma, GlaxoSmithKline, Pfizer Global R&D and Novartis Pharmaceuticals leading groups of scientists in development and validation of analytical methods supporting new drug development through product life-cycle.
Ivone Takenaka, PhD is an Associate Director in Global Regulatory Affairs-Chemistry, Manufacturing and Controls Department, and is the Latin-America-Canada Regional Expert at Bristol-Myers Squibb Co. She earned her Ph.D. and Master’s Degree in Molecular Biology & Biochemistry from the University of Connecticut, CT; and Master’s Degree in Quality Assurance and Regulatory Affairs from Temple University, PA. She held different positions in Drug Metabolism & Pharmacokinetics, and Oncology Drug Discovery at Bristol-Myers Squibb prior to moving to regulatory affairs. She held an Associate Director position in Corporate Quality & Regulatory Affairs at Abbott Laboratories.
John A. Castoro, PhD is an Associate Director in the Analytical and BioAnalytical Department at Bristol-Myers Squibb Company. He earned his Ph.D. in analytical chemistry at the University of California, Riverside. For the last 20 years his areas of focus include analytical method development in support of drug substances and products as well as impurity structure elucidation. He presently leads a group that supports the drug development from early development to approval