The topic of LER, or “Low Endotoxin Recovery” has dominated endotoxin discussions since 2013. What is LER? The term was coined to describe an LAL assay interference (inhibition) that was observed in an undiluted monoclonal antibody formulation containing a chelating buffer and polysorbate (Chen and Vinther, 2013). LER has raised questions regarding patient safety and the validity of the compendial Bacterial Endotoxins Test in detecting low levels of endotoxin contamination in biological products. Since the initial report of LER, the United States Pharmacopeia, General Chapters- Microbiology Expert Committee (EC), as the steward for Bacterial Endotoxins Test <85> (USP, 2016a), has taken a keen interest in understanding the implications of the LER phenomenon. This article is the first of two in this supplement that describes current USP thinking. The second is “NOE: A New Endotoxin Standard Proposed by USP” by Radhakrishna S. Tirumalai, PhD.
It is important that the lexicon used in the following discussion be established to define the terms used, because the lack of a shared understanding of the vocabulary associated with LER has added to the confusion on the subject. It is generally agreed that “endotoxin” is defined as a component of the outer cell membrane of Gram negative bacteria, whereas the term, “lipopolysaccharide” (LPS), and the Lipid A portion in particular, is defined as the specific component of the endotoxin complex that evokes pyrogenic responses in mammals and initiates signal generation in LAL tests. LPS is an amphipathic molecule with a hydrophobic portion (Lipid A) that is embedded in the cell membrane and hydrophilic (O-antigen) portion that is exposed to the extracellular environment
LPS can be extracted from the cell wall of Gram negative bacteria in a laboratory by any number of methods and further purified for use as an analyte for research or as a calibration standard for the various LAL assays. Extraction and purification strips the LPS of its associated cell wall components and the now “naked” amphipathic LPS can stick to solid surfaces and will take on various aggregate forms in aqueous solution including micelles, ribbons, and other conformations not found in nature. USP’s Reference Standard Endotoxin (RSE) and all of the commercially available control standard endotoxins (CSE) currently associated with LAL test kits are, in fact, extracted and purified preparations of “naked” LPS from various strains of Escherichia coli. The emphasis on vigorous mixing of RSE and CSE in an LAL test is to assure the proper resuspension of the lyophilized material, prevent adsorption of LPS to the surface of vessels, and optimize the LPS aggregation state for testing. Too little or too much aggregation will impact LAL test controls.
Endotoxin contamination in a product can only arise from the introduction and proliferation of Gram negative bacteria via the raw materials (including water) or during the manufacturing process. During the natural bacterial cell cycle, pieces of the cell membrane are pinched off from the growing cell forming extracellular spherical vesicles, or “blebs”. Once released, these bleb float freely in the extracellular environment without the same aggregation patterns seen with purified LPS. Electron micrographs clearly demonstrate the physical and structural differences between the natural endotoxin blebs and purified LPS in aqueous solution (Brogden and Phillips, 1988).
It is the position of the EC, that significant biochemical and physical differences between native endotoxins and calibration standards exist. These differences make the use of RSE and CSE scientifically questionable as de facto “like for like” or “worst case” surrogates for natural endotoxin contamination in hold time and other endotoxins recovery studies.
The possibility of a false negative LAL test due to LER coupled with a documented pyrogenic response due to the presence of contaminating endotoxin is clearly a patient safety concern. The “LER formulation” of a therapeutic protein, chelating buffer and polysorbate has been used in the biotechnology industry for almost 30 years and presumably this LER phenomenon has also existed for that same period of time. While there is always the possibility of a pyrogenic reaction in any given patient as a result of the infusion process or perhaps the mode of action of the therapeutic protein itself, had a batch of product been released with high levels of contaminating endotoxin, one would expect to see a notable cluster of adverse events related to that lot, resulting in a recall or other action. However, no such reports exist either in the scientific literature or among reports on FDA’s website because much improved process controls coupled with valid LAL testing for in process and release testing have done a good job in keeping the supply of these biologics safe. In the absence of Gram negative contamination, interference with the BET assays is an analytical issue not a public health one.
Concurrent rabbit Pyrogen Tests (RPT) and LAL assays in an LER formulation hold time study using RSE and a variety of laboratoryderived bleb preparations (Enterobacter cloacae, Ralstonia picketti, Escherichia coli, Pseudomonas aeruginosa and Serratia marcescens) as spiking analytes have been conducted. At the outset of the study (T=0), all of the spiked preparations, including the RSE, caused similar pyrogenic responses in the rabbits and each of the spikes was fully detected in the LAL test. After a twenty four hour hold, all of the bleb preparations were still reactive in both the RPT and were fully recovered in LAL assays, but the RSE was nonreactive in both the RPT and LAL, which is the LER response (J. Dubczak as reported in Bolden, et al, 2015). These data suggest that LER is not an issue of LAL test validity, but rather an issue of the choice of spiking analyte used as a control as well as the experimental design for hold time studies (Bolden, et al, 2014; Platco, 2015; Bolden, et al., 2015; Dubczak, 2016).
Reports from companies with LER affected products indicate these formulations have all met the requirements of USP 85 Test for Interfering Factors (inhibition/enhancement) and therefore meet the compendium’s validity criteria for routine testing. So, how can a product with a valid BET assay per USP <85> also have LER interference?
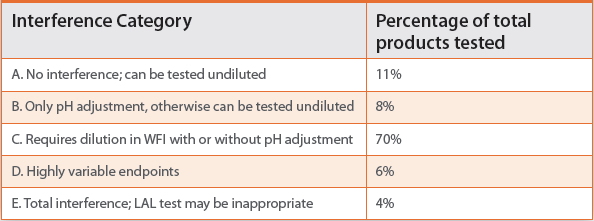
The harmonized Bacterial Endotoxins Test chapter instructs the analyst to add a known amount of RSE to each tube in a series of dilutions or preparations of product at an activity level of 2 lambda for gel clot assays and the midpoint of the standard curve for quantitative assays. Each of these “spiked” dilutions is tested individually using the referenced assay, and the analyst is instructed to identify a dilution or a sample preparation method that allows for the quantitative (kinetic or endpoint assay) or qualitative (gel clot) recovery of RSE activity. This control is commonly known as the Positive Product Control, or PPC (USP, 2016a). We know from experience and the published literature that most products, when tested undiluted, inhibit the LAL test, meaning that the interference will underestimate PPC recovery. Table 1 summarizes data from a study published by Christine Towhy and her colleagues at FDA in 1984 suggesting that fully 89% of products tested by the FDA laboratories at the time interfered with recovery of activity when tested undiluted. These data were collected at a time when most parenteral products were small molecule drugs with relatively simple formulations. LER associated products, like 70% of products in the 1984 study, fall into Category C, and require dilution in Water for BET to overcome interference for USP <85>.
Table 1. Summary results: Twohy, et al (1984)
The current compendial test does not require the analyst to “spike” undiluted product with RSE and demonstrate that the spike dilutes with the product. However, in 2012, FDA withdrew a long standing Guideline on LAL (FDA 1987) and replaced it with a series of questions and answers (FDA 2012). Question 3 addressed the importance of sample storage and handling, and FDA indicated that “assayable” endotoxin should be stable during storage and handling. However, the term, “assayable endotoxin” was never defined. Was FDA asking for assurance that a lot of product naturally contaminated with endotoxin blebs is stable in its activity or were they asking for prospective spiking studies using calibration standard (RSE or CSE)?
A closer reading of the response to Question 3 finds a disclaimer from FDA that states, “…purified endotoxins might react differently from native sources of endotoxins.” Data generated almost 50 years ago from two different laboratories (Ribi, et al, 1966; Hannecart- Prkorni, et al, 1973) and more current data reported by Mueller and coworkers (2004) and Tsuchiya (2015) suggest that the normal LPS aggregate conformations found in aqueous solutions tend to disaggregate in the presence of chelators and surfactants (the “LER formulation”), and as the aggregates get smaller, approaching the monomer state, the biological activity decreases as well. Acknowledgement of FDA’s disclaimer coupled with a review of the literature would have predicted the interference observed with LPS in the LER formulation.
In a 1979 report, Carmine Mascoli and Marlys Weary, working at Travenol Laboratories (now Baxter) reported on the concurrent LAL and rabbit pyrogen testing of eighty seven (87) regular production lots of a biological product, 25% albumin. Their focus was on understanding the correlation between LAL measurements and rabbit fever responses, including the impact of test method interference, to ascertain the validity of the LAL test in predicting rabbit pyrogen failures. Their data demonstrated the following:
74 of 87 lots (85%) passed both the rabbit Pyrogen and LAL testing; None of the lots was found to pass the LAL test yet fail the rabbit test, meaning that no false negative LAL tests were observed. 11 of 87 lots (12.6%) failed both the rabbit and LAL tests 2 of 87 lots (2.4%) failed the LAL test but passed the rabbit pyrogen test.
The results from these “real life” contaminated products coupled with almost 40 years of successful LAL testing and the recent RPT/ LAL data as reported in Bolden, et al (2015) strongly suggest that the compendial BET assays are not only valid, but are good predictors of pyrogenicity and patient safety.
A number of researchers have reported that the LER effect in these biological products can be mitigated when the spiking analyte is an endotoxin bleb preparation rather than a surrogate purified LPS preparation (Bowers and Tran, 2011; Bolden, 2014; Bolden, et al, 2015; Platco, 2015; Dubczak, 2016 and unpublished communications to USP). However, concerns have been raised that inconsistencies in the preparation of these bleb solutions may add considerably to the variability of test results.
In a separate initiative, the EC is developing a series of informational chapters on depyrogenation. Chapter <1228.5>, “Endotoxin Indicators” acknowledges the utility of an endotoxin bleb preparation as a surrogate for natural contamination in prospective depyrogenation studies for product streams and has provided some guidance in that chapter for the generation of such preparations (USP 2016b). The confluence of these two applications for an endotoxin bleb standard, LER testing and endotoxin indicators for product stream depyrogenation, has led USP to propose a new “naturally occurring endotoxin” (NOE) standard preparation comprised of native endotoxin. For a detailed discussion of this proposed standard, see Part II of this series.
Summary
After careful consideration, the EC has determined that there are no data in the trade literature, in the peer reviewed scientific literature, or on FDA’s website that would suggest that LER poses a patient safety concern or that the current <85> is invalid for the routine testing and release of parenteral products.
Each of the “LER” biological products has met the requirements of USP <85> and none has been recalled for endotoxin contamination, suggesting that the improved manufacturing controls and valid LAL assays are keeping the supply of parenteral biologics safe.
It has been known for at least 50 years that chelators and polysorbate used in “LER formulations” cause the lipopolysaccharide aggregates to dis-aggregate into biologically inactive monomers suggesting that the LER interference is more a function of the analyte that is chosen for a study rather than an LAL assay validity issue.
Published data indicate that under the right experimental conditions, the “LER” effect observed with RSE or CSE may be overcome by using native endotoxin blebs as the analyte.
Researchers have long used LPS as a surrogate analyte for endotoxin blebs in their work, and many have held that the terms, “endotoxin” and “LPS” are interchangeable. After all, both are capable of evoking pyrogenic responses in mammals and both are capable of initiating the LAL cascade. If the LER controversy has taught us anything, it’s provided reproducible data to support what we’ve always known – that LPS and endotoxin, while perhaps evoking similar responses are very different chemically, structurally, and physically and are subject to different influences from the environment in which they exist. Current published research suggests that the cell wall components that surround the LPS molecules in bleb preparations may afford some protection against interferences such as LER that may affect the chemistry of spiking analyte rather than the test reagent. It is therefore important to provide clarity when using the term, “endotoxin.”
Literature Cited
- Bolden, Jay, Mark E. Clarebout, Matthew K. Miner, Marie A. Murphy, Kelly R. Smith, Rob E. Warburton. 2014. Evidence Against a Bacterial Endotoxin Masking Effect in Biologic Drug Products by Limulus Amebocyte Lysate Detection. J. Parent. Sci Tech. 68(5): 472-477
- Bolden, Jay, Cheryl Platco, John Dubczak, James F. Cooper, Karen Zink McCullough. 2015. The Use of Endotoxin as an Analyte in Biopharmaceutical Product Hold-Time Studies. United States Pharmacopeia: Stimuli to the Revision Process 41(5).
- Bowers, Kim and Lynn Tran. 2011. Creation of an in-house Naturally Occurring Endotoxin Preparation for Use in Endotoxin Spiking Studies and LAL Sample Hold Time Analysis. American Pharmaceutical Review. September/October. Pages 92-97
- Brogden, K.A., and M. Phillips. 1988. The Ultrastructural Morphology of Endotoxins and Lipopolysaccharides. Electron Microsc. Rev. 1: 261-277
- Chen, Joseph and Anders Vinther. 2013. “Low Endotoxin Recovery in Common Biologics Products.” Presented at the 2013 PDA Annual Meeting, Orlando Florida.
- Dubczak, John. 2016. The Great LER Debate. Outsourced Pharma. http://www.outsourcedpharma.com/doc/the-great-ler-debate-0001
- Hannecart-Pokorni, Elenora, Daniel Dekegel, Freddy Depuydt. 1973. Macromolecular structure of Lipopolysaccharides from Gram negative Bacteria. Eur. J. Biochem. 38: 6-13
- Mascoli, Carmine and Marlys Weary. 1979. Applications and Advantages of the Limulus Amebocyte Lysate (LAL) Pyrogen Test for Parenteral Injectable Products. In: Bioomedica Applications of the Horseshoe Crab (Limulidae). Alan R. Liss, Inc. NY, NY. Elias Cohen, Editor.
- Mueller, M., B, Lindner, S. Kusumoto, K. Fukase, A.B. Schromm and U. Seydel. Aggregates are the Biologically Active Units of Endotoxin.” J. Biol. Chem. 279(25): 26307-26313.,
- Ribi, E., R.L. Anacker, R. Brown, W.T. Haskins, B. Malmgren, K.C. Milner, and J.A. Rudbach. 1966. Reaction of Endotoxin and Surfactants. J. Bacteriology. 92(5): 1493-1509)
- Tsuchiya, Masakazu. 2014. Possible Mechanism of Low Endotoxin Recovery. American Pharmaceutical Review. 17(7).
- Twohy, Christine W., Anthony P. Duran, Terry E. Munson. 1984. Endotoxin Contamination of Parenteral Drugs and Radiopharmaceuticals as Determined by the Limulus Amebocyte Lysate Method. J. Parent. Sci. Tech. 38(5): 190-201
- United States Food and Drug Administration 2012. Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm310098.pdf
- United States Food and Drug Administration 1987. Guideline on Validation of the Limulus Amebocyte Lysate Test as an End Product Endotoxin Test for Human and Animal Parenteral Drugs, Biological Products, and Medical Devices.
- United States Pharmacopeia. 2016. <85>, “Bacterial Endotoxins Test”
- United States Pharmacopeia. 2016. <1228.5>, “Endotoxin Indicators
Karen Zink McCullough is principal consultant at MMI Associates, a consulting firm focusing on Quality System development and pharmaceutical microbiology. Ms McCullough is nationally and internationally known for her work in the Bacterial Endotoxins Test (BET), and is a frequent speaker, instructor author on such topics as BET, GMP, Metrics, Risk, and pharmaceutical microbiology. Her credits include editing two books on Microbiology and BET, authoring 26 book chapters and 19 published articles. She is an elected member of the USP Expert Committee, Microbiology General Chapters and is a US delegate to ISO TC209, Working Group 2, “Biocontamination”. Ms. McCullough received her BA degree in Bacteriology from Rutgers University and her MS in Molecular Biology from the University of Oregon.