The Low Endotoxin (LER) and Lipopolysaccharide Recovery (LLR) phenomena have been debated as a Limulus Ameobocyte Lysate (LAL) test validity issue peculiar to therapeutic monoclonal antibodies for over three years. The FDA division reviewing Biological License Applications has requested that firms provide data which demonstrate the stability of assayable endotoxins as required in the FDA issued Guidance for Industry Pyrogen and Endotoxins Testing: Questions and Answers, No. 3.1 In 2013, scientists from Genentech reported the swift loss of measurable LAL activity when purified lipopolysaccharide (LPS), used as endotoxin activity standards was added directly into a citrate/ polysorbate monoclonal platform.2 The LPS was added as a surrogate for bacterial endotoxin contamination which the product formulation may encounter. A conclusion was reported to the FDA and shared with the PDA’s 2013 Annual Conference participants stating that the assay for bacterial endotoxins was inappropriate for its intended purpose; this despite meeting the compendial criteria for a valid assay.
A crucial factor in the recovery of the activity of any surrogate contaminant when directly added into product matrices is the relevant preservation of the analyte molecule’s size, primary structure,conformation and charge.3 Endotoxins are amphillic molecules and form multiple aggregates above a certain concentration (critical aggregation concentration); hydrophobicity, size, structure and charge all contribute to the relative proportion of multimeric aggregates or monomers. Secondary standards such as control standard endotoxin (CSE) and the primary standard reference standard endotoxin (RSE) are chemically purified LPS molecules. Controlled preparation of both CSE and RSE involves phenolic extraction to prepare an LPS relatively free of biologically active proteins, polynucleotides, etc.4 This treatment imparts chemical and structural characteristics quite distinct from native or natural occurring endotoxin (NOE) counterparts. Despite chemical and structural differences both LPS and NOE molecules share an ability to activate limulus, monocytes, and provoke an innate pyrogenic response in rabbits. Other attributes such as molecular size, conformation, aggregation state, chemical stability, and associated uniformity are very fundamentally distinct. Therefore, because of different chemistries inherent to RSE, CSE and NOE their stability characteristics and liability to chemical influences imparted from buffer matrices are distinctly different. The terms LPS and NOE have erroneously been used interchangeably with respect to attributes other than pyrogenic function.
The purpose of the Bacterial Endotoxin Test (BET) test is to determine lipid A (pyrogenic) activity. Indeed, it is lipid A that is responsible for the biologic activity of LPS in both in vitro and in vivo test systems; and coining the term the endotoxic principle of endotoxin.5 The test is quite effective and accurate at measuring lipid A activity providing lipid A presents itself available for detection. Data accumulated over the past three years demonstrate that LPS activity is not always stable, but may rather diminish by being ‘blanketed’ or ‘masked’, or is destroyed when directly encountering chelating product buffers. Purified LPS is not an authentic substitute or surrogate for Gram negative NOE contamination activity due to inherent differences in chemical stability, molecular size, and aggregation state. In comparison to purified LPS. NOE is not prone to a loss of activity (LAL and RPT tests) when directly encountered by most buffer platforms including citrate/polysorbate formulations. Retention of this activity is attributable to the chemical and molecular stability of NOE. NOE has thus far been recovered from a wide range of buffer platforms including citrate/polysorbate formulations at both room temperature and cold storage conditions for extended periods of time. The citations are numerous and the data are compelling. An article written as a Stimuli Article to the Pharmacopial Forum contains many references.6 Rare cases have been reported where all LAL activity, whether purified LPS or NOE, is also reduced or destroyed. Industry debates continue with burgeoning discussion on an as yet unsubstantiated hypothetical concern over patient safety associated with any Gram negative impurity which has no pyrogenic activity. This is a topic for another discussion. Here we shall focus on pyrogenic activity and solely use the terms Low Lipopolysaccharide recovery (LLR) for scenarios where material other than NOE is used. In scenarios where NOE is involved we use the term Low Endotoxin Recovery (LER). The authors believe that a correct, accurate and consistent use of terminology is a necessity when considering complex phenomena. As more data is presented and published, greater clarity concerning the “masking” phenomenon will occur permitting us to truly address if LER is really just a mechanism by which LPS activity is decreased or lost and if this “masked” LPS can be uncovered and reactivated’.
The most controversial aspect of LER discussions has involved the appropriate choice of endotoxin molecules used as an appropriate surrogate contaminant for hold time studies. Such hold time studies evaluating the potential for impact of Gram negative bacteria derived endotoxin to adulterate a drug product. One school of thought favors laboratory prepared and purified LPS molecules because they might be argued as “worst case.” However, it is indeed a truism that LPS does not exist naturally and would not be encountered unless artificially prepared in a laboratory. Fundamentally LPS only shares Lipid A activity with NOE molecules, genuinely representative of Gram negative derived endotoxin which a drug substance may actually encounter. Research and data informs us that LPS can disaggregate to the point where it is no longer biologically active.7
For any assay to measure endotoxin activity from either purified LPS or NOE molecules, the active lipid A portion of the molecule must present itself to the activation or detection mechanisms. This is true for any of the LAL techniques, Monocyte Activations tests, rFC LAL techniques and the newer ELISA assays. All of these assays are subject to LLR phenomenon because the conditions under which this is encountered are not method or technique specific. Therefore all methods require some type of sample pretest preparation to allow the lipid A inherent to LPS to become available to the detection system.
The presentation of lipid A may be highly dependent upon the mechanism for LLR in the first place. Thus far industry has focused on the masking mechanism.8,9 Others have proposed that the monoclonal antibody itself may attach to the LPS molecule rendering the lipid A inactive or inaccessible for keying to receptors for any of the detection systems. When purified LPS molecules are used as surrogate analytes, the masking is likely linked to the smaller size of the chemically purified molecules. In contrast, larger endotoxin constituent fragments of Gram negative cell wall debris are mostly unaffected; the size and chemistry of the fragments preserving the accessibility of the lipid A moiety.

Industry has been requested by the FDA to use LPS as a surrogate contaminant. Firms have to discover a sample pretest preparation which will detect the activity of the lipid A after it has been in direct contact with the product matrix over a period of time. Both simple and complicated sample preparations to unmask, reactivate, and/or re-aggregate the smaller LPS molecules to an active form have been investigated. Choosing a demasking technique can be difficult and complicated; necessitating an understanding of the mechanism that has caused the failure to detect LPS and/or NOE in the first place. The sample matrix may chemically destroy the activity of lipid A both LPS and NOE molecules if aggressive enough; in these cases, restoring biological activity may not be possible. It is recommended to evaluate simple solutions before reaching into the toolbox for elaborate and costly sample pretest preparations. Many of the more elaborate sample pretest preparations require extensive sample manipulations simultaneously bring added risk of accidental contamination.
The authors propose the following first steps when LLR is encountered with mAb formulations, recognizing that these are merely suggestions of proven merit, yet may not resolve all LLR issues.
Where feasible use kinetic LAL methods. The kinetic methods are most likely to detect LLR because the kinetic detection methods determine a ‘race to a finish line’ known as the onset time. In comparison, the gel clot assay may not detect LLR as easily if at all. As an endpoint test, slower reacting tubes are not identified as the test results are not observed until one hour. The slower reacting tubes will have formed a gel, the slower gel formation not being observed. The gel clot assay also has very high variability and slight inhibitory conditions may be missed.
Investigate if the monoclonal antibody itself is causing lower biological activity of CSE and/or NOE by evaluating formulations containing either the active biological molecule or a placebo product. Most importantly, it is imperative to demonstrate that a sample pretest preparation developed to restore lipid A activity does not affect NOE activity. In reality if a product formulation encounters endotoxin it will be in the form of NOE; representative of Gram negative bacterially derived cell wall and not purified and chemically extracted LPS.
Another avenue to investigate is to test multiple lots of product treated with or without the pretest product preparation methodology to discover if any “stealth” endotoxin activity is present. If endogenous NOE is present, the stability of assayable endotoxins can be achieved by collecting data over time during stability studies to assure that the endotoxin limit is never exceeded. Reviewing the FDA issued Guidance for Industry Pyrogen and Endotoxins Testing: Questions and Answers, No. 3, we can clearly see that this was the original intended purpose. Also, there has never existed a requirement to add surrogate CSE in addition to an already present NOE to determine if LLR conditions exist.
LLR experimental design is profoundly important and small changes may significantly alter the outcome. Ensure that the enhancement/inhibitions properties of the product matrix are well defined where positive product controls (PPC) are 50-200%. The non-interfering dilution (or greater) is used for testing product contaminated with LPS. Place an appropriate amount of product into individual glass vials for each planned test day. Prepare LPS (CSE or RSE) at a stock concentration of approximately 1000 EU/mL or higher so that one does not dilute out the product matrix with surrogate analyte. Contaminate the product so that the resultant activity after test dilution is at approximately the midpoint to ¾ of the standard curve range. The volume of contaminant should not exceed 1% of the total volume of product in the vials. Prepare the same set of artificially contaminated purified water controls (PFW). Add the LPS contaminant to the vial for the last day of the holding period first, then adding the LPS contaminant to the remaining vials in sequence from longest hold time to zero hold time. In this manner, all testing can occur on the same day using the same standard curve thus avoiding the introduction of unnecessary assay variables. Store the samples including the PFW at 2-8°C (recommended storage condition for monoclonal antibodies and also for CSE standards).
Review the data and interpret the results carefully. The water controls should be used to determine % activity recovery from the samples. Theoretical values may not be accurate; the theoretical activity value will only be valid if one has contaminated the samples with CSE from the same CSE:Lysate paired lot used for testing. The theoretical value will be inaccurate unless the contaminant CSE was standardized or measured prior to use with the reagents from the method (paired standard and lysate). A value of not more than a 50% loss of activity when compared to the water control is generally been accepted as evidence of the absence of LLR and a reactivation of at least 50% CSE is satisfactory for sample pretest preparations.
What if the data indicate LLR, currently defined as more than 50% loss of CSE activity and the sample matrix is citrate or phosphate buffer containing with polysorbate?
The LPS masking mechanism is caused by the chelation effect of the buffer and the aggregation state of the molecules.8,9 While each therapeutic monoclonal antibody formulation is individual in terms of structure and chemistry, the data presented below demonstrates success has been achieved at resurrecting the CSE activity by using commercially available cation replacement/ TRIS buffer Stock cation buffer (0.5M MgS04, 1M TRIS) was diluted in pyrogen free water to be used as a sample preparation diluent. The concentration of the cation buffer has to be optimized for each lysate vendor and each method/technique prior to use. Otherwise, inhibition of the test detection mechanism may occur. For instance, we have determined that a concentration of 100mM TRIS, 50mM MgS04 was the non-interfering concentration of cation buffer to unmask CSE/LPS from a kinetic turbidimetric method supplied by vendor A. However, a kinetic chromogenic method from vendor B was inhibitory at the same cation buffer concentration. The noninterfering concentration of cation buffer was 25mM TRIS, 12.5mM MgS04 and required a higher product dilution. The data from this study is similar to Table 1.
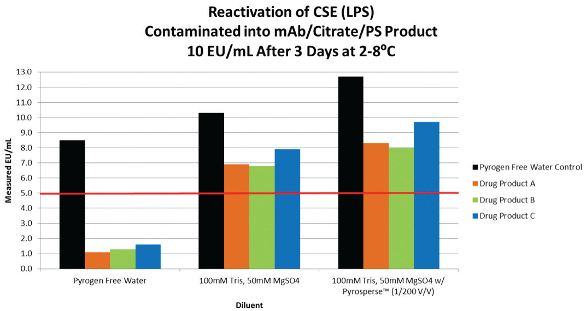 Table 1.
Table 1.Dilute the masked LPS product in the cation diluent and allow the dilution to rest at room temperature for 10 minutes, vortexing briefly every few minutes. We have resurrected the CSE activity in our monoclonal citrate/ PS based platform with about an 80-90% recovery. (Table 1) We also used a dispersing reagent added directly into the contaminated sample at a concentration 1/200 (v/v) prior to diluting the samples in the cation buffer. The recovery of masked LPS was not significantly better with dispersing reagent, but demonstrated no disadvantages to its use. It is very important to store the sample during the masking process at 2-8°C as storage at room temperature will speed up the masking process, but loss of activity was found to be irreversible (Table 2). The contaminated water controls were unaffected by the sample treatment process (Table 3). NOE activity was also evaluated through this same sample pretest preparation. The NOE activity (the NOE was derived from Enterobcter cloacae, at a stock activity value of 768 EU/mL) was unaffected and stable whether diluted in PFW, cation buffer, or treated with dispersing reagent. The same success was achieved by another vendor kit using kinetic chromogenic testing (Tables 4 and 5). Another interesting observation during the course of these studies was that the LPS masking was never 100% complete regardless of the amount of CSE added. There was always a small but proportional amount of activity which would not mask. A probable explanation is that the chemically purified LPS standards is heterogeneous and contains a portion of molecules that are more stable and are more NOE-like in structure.
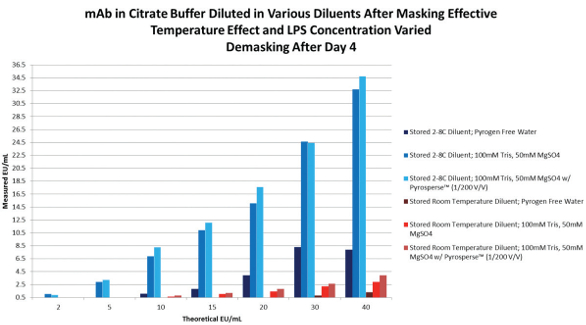 Table 2.
Table 2. Table 3.
Table 3. Table 4.
Table 4.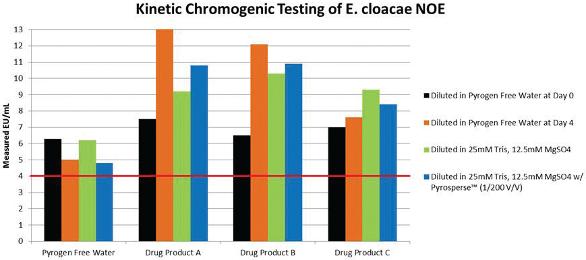 Table 5.
Table 5.The USP microbiology expert committee has proposed the use of a well characterized NOE preparation to be used for depyrogenation studies as well as for stability of assayable endotoxin/hold time studies. Preparations similar to this have been reported in prior publications.10 While secondary standards are not required to be purified LPS, the use of LPS/CSE standards will remain. The NOE preparation would be standardized (NOE:RSE) in terms of activity in a similar manner as the CSE are standardized as secondary standards against the primary RSE standard. Acceptance of a more robust artificial surrogate contaminant will prevent spurious recovery data concomitant with the use of the chemically purified LPS. The NOE will more authentically represent endotoxin derived from Gram negative microorganisms and resolve the experimental design flaw of using a surrogate contaminant for which the attributes of size, conformation, and susceptibility to chemical interference/ destruction are irreconcilable with endotoxin that a product may actually encounter.
There are currently three organizations with ‘LER task forces’ working for a resolution of this topic. It has been most helpful that the FDA participates in these discussions. The Pharmaceutical Drug Association, BioPhorum Operations Group, and the 35+ year industry LAL User’s Group sponsored by the Pharmaceutical Microbiology Forum are all committed to assuring that the bacterial endotoxins test assures patient safety. It appears that the only interchangeable attribute between the molecular types is lipid A activity as a measure of pyrogenic activity. With or without these sample pretest preparations, the compendia USP/ EP/JP bacterial endotoxins tests are now and continue to be suitable for their intended purpose which is testing product safety for Gram negative bacterial endotoxin pyrogenic activity.
References
- Food and Drug Administration. 2012. Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers.
- Chen J, Vinther A. Low endotoxin recovery (LER) in common biologics products. Parenteral Drug Association Annual Meeting, Orlando, 20131
- Schromm AB, et al. The Charge of Endotoxin Molecules Influences Their Conformation and IL-6-Inducing Capacity. J. Immunology, 1998, 161 (10), 5464-5471.
- Rudbach JA, Akiya J, Elin RJ. Preparation and properties of a national reference standard endotoxin. J.Clin. Microbiol. 1976; 3:21-25.
- Rietschel, E. T., H. Brade, L. Brade, K. Brandenburg, U. Schade, U. Seydel, U. Zähringer, C. Galanos, O. Lüderitz, O. Westphal, H. Labischinski, S. Kusumoto, T. Shiba. Lipid A, the endotoxic center of bacterial lipopolysaccharides: relation of chemical structure to biological activity. Prog. Clin. Biol. Res. 1987. 231: 25
- Bolden J, Platco C, Dubczak J, Cooper J, McCullough KZ. The use of endotoxin as an analyte in biopharmaceutical hold time studies. Published in Pharmacopeial Forum, 2015.
- Mueller, M. et al. “Aggregates are the Biologically Active Units of Endotoxin.” The Journal of Biochemistry, 279, 26307-26313, 2004.
- Tsuchiya M. Possible mechanism of low endotoxin recovery. American Pharmaceutical Review. 2014;17(7):18-23.
- Reich J, et al., Masking of endotoxin in surfactant samples: Effects on Limulus-based detection systems, Biologicals (2016), http://dx.doi.org/10.1016/j.biologicals.2016.04.012
- Bowers K, Lynn T. Creation of an in-house naturally occurring endotoxin preparation for use in endotoxin spiking studies and LAL sample hold time analysis. Am Pharm Rev. 2011;14(6):92–97.