Small Molecule Pharmaceutical Sciences
Small Molecule Pharmaceutical Sciences Department
Abstract
This review focuses on the analytical challenges of chromatographically characterizing sulfonate salts/esters, hydrazine functionalities, amines, boronate esters/acids, aldehydes, and sulfonate acid/esters, and acyl (acid) halides used in the synthesis of pharmaceutical drug substances. Special focus is placed on stability, degradation, and achieving the low level sensitivity required for genotoxic impurity analysis. Final pharmaceutical drug substances are designed to be stable to meet shelf life requirements, survive pharmaceutical processing into drug products, and intact delivery through the GI and/or bloodstream to their sites of action. However, the building blocks of these APIs are not constrained by the same stability requirements and may require special considerations to be accurately analyzed.
Introduction
Reactive molecules are the building blocks of synthetic organic chemistry and can be found throughout the pharmaceutical industry in Suzuki Couplings, antibody drug conjugations, etc. Their reactive nature drives syntheses, thereby allowing the catalysts, reagents, and reaction conditions to focus on the selectivity of the reaction. Even in the coupling of two non-reactive molecules, one of the components will often be converted to a reactive species in situ prior to formation of the bond between the two molecules. In the pharmaceutical industry, reactive molecules are often utilized as starting materials and isolated intermediates in the synthesis of the complex, selective, and biologically active new small molecule pharmaceuticals. As commercial processes are developed, reactive molecules are often shifted from starting materials and isolated intermediates to un-isolated intermediates; however, the control of these un-isolated intermediates remains critical to the robustness, cost, quality, safety and environmental impact of the synthetic route.1 Additionally, some molecules that are deemed unreactive from a synthesis perspective can be considered reactive in the analytical laboratory due to the necessary exposure to water and/or air during sample preparation and analysis.
Accurate analysis of these reactive molecules is key to developing, monitoring, and controlling pharmaceutical syntheses from early development through commercial manufacturing. The data obtained from these analyses are used to set purchasing specifications, ensure proper charging for reaction stoichiometry, monitor the progress of the reaction, study yield and mass balance of processes in designed experiments, and evaluate purity of the reactive products.
Accurate and sensitive analysis of reactive molecules poses a challenge when decomposition is encountered during sample preparation and testing. These molecules can decompose by various mechanisms including: oxidation, reduction, hydrolysis, polymerization, condensation, elimination, substitutions and isomerization.2,3 Further, the inherent reactivity of these molecules often raises concerns regarding their reaction with DNA and consequently concerns of mutagenic potential. This mutagenic potential is of increasing concern to health authorities often requiring control of potentially mutagenic compounds to ppm/ ppb levels.4,5
Direct spectral analysis by techniques such as quantitative NMR (nuclear magnetic resonance)6 and vibrational spectroscopy (mid-IR, near-IR and Raman)7 is often used to minimize sample preparation and decomposition of reactive species. While direct spectral analysis is accurate and minimizes decomposition, sensitivity and selectivity are limitations, especially in the presence of complex sample matrices or when ppm-level sensitivity is required. This paper focuses on the techniques that are widely used in the pharmaceutical industry,namely, chromatographic separations followed by inline detection, and describes the considerations necessary to apply these techniques to reactive molecules.
Overview of Commonly Used Analytical Technologies
Direct analysis by reverse phase liquid chromatography (RP-HPLC) is the preferred separation technique in the pharmaceutical industry due to its ability to resolve complex mixtures with gradient elution, the extensive selection of available stationary phases available commercially, and its compatibility with a range of detectors including ultraviolet/visible (UV/VIS), mass spectrometry (MS), corona aerosol detector (CAD), evaporating light scattering detector (ELSD), etc. However, RP-HPLC ‘s utility may be limited when analyzing molecules that are reactive to water, silanols, or modifiers in the mobile phase such as acids, bases and buffers.
Some reactive molecules, such as boronate esters8,9 are amenable to direct analysis using RP- HPLC conditions when special considerations are taken. However, in the majority of cases, accurate and sensitive analysis of reactive molecules requires alternate methodologies.
Derivatization has long been used to facilitate analysis of reactive molecules as derivatization serves to stabilize the molecule while often affording the opportunity to increase method sensitivity. Common derivatizations include alkylation, silylation, acylation, and chiral derivatizations.10 Strategies for derivatization of a myriad of compounds have been developed for both HPLC and GC analysis.11,12 While derivatization is a valuable technique, the analytical chemist may wish to avoid it to minimize sample preparation time and to address concerns over incomplete conversions or side reactions.
Normal phase chromatography (NP-HPLC) and supercritical fluid chromatography (SFC) have also been used to analyze molecules which are not stable in the aqueous phase required for reverse phase chromatography. In some cases, mobile phase modifiers such as acids and bases may also be eliminated in NP-HPLC and SFC, thereby addressing another potential route of degradation. Normal phase liquid chromatography is not without its drawbacks, however; sensitivity of late-eluting peaks may be inadequate due to bandbroadening13 as NP-HPLC is not amenable to gradient elution. Further, isocratic elution lacks the resolving power required for complex mixtures. SFC addresses this limitation, thereby allowing the analytical chemist to use normal phase solvents with gradient elution. Although SFC is once again gaining rapid adoption, this separation platform is not as widely available as HPLC, and therefore RP-HPLC may still be preferred to SFC. It should be noted that both NP-HPLC and SFC are limited to molecules that can be dissolved in normal phase solvents.
Finally, gas chromatography (GC) allows for both direct analysis and the analysis of derivatized species. Multiple GC detectors, including MS, thermal conductivity detector (TCD), and flame ionization detection (FID) are available, with the latter two’s response reflective of concentration across analytes. While water can be eliminated from the sample prep, thereby enabling analysis of water-sensitive compounds, GC remains limited to volatile and thermally stable molecules such as sulfonate esters14 and allyl chlorides.15
No single methodology is appropriate for characterization of all species of reactive molecules, but by considering the properties of the molecule and their compatibility with a wide range of analytical techniques, the modern analytical chemist can develop accurate and sensitive methods for all but the most reactive species.
Strategy for Reactive Molecules Analysis
This paper presents several case studies where accurate and sensitive analytical methods have been developed to analyze reactive molecules. Table 1 outlines various classes of reactive molecules used in the pharmaceutical industry, their synthetic utility, mutagenic potential, and analytical strategies for their characterization.
Table 1. Synthesis Utility, Mutagenic Potential, and Analytical Strategy for Various Reactive molecules.
Analysis of Acids
Acids, such as carboxylic and sulfonic acids, are widely used in the synthesis of pharmaceutical compounds. They may be a part of the synthetic scheme itself, introduced to form a salt, used as a catalyst, or charged directly to the reaction mixture to control the pH. Additionally, acids may form as a byproduct, such as during an elimination reaction.
Typically, acids are analyzed using traditional HPLC methods with little concern for their reactivity. However, use of protic solvents (methanol, ethanol, etc.) should be avoided as a sample diluent or during analysis due to potential esterification reaction with the analyte unless derivatization of the acid with the alcohol is a desired outcome.
If protic solvents are necessary to achieve an adequate separation, the short term stability of the acid in the mobile phase should be confirmed off-line, taking into consideration HPLC parameters that could enhance the esterification reaction such as elevated column temperatures.
Certain sub-classes of acids represent significant analytical challenges during analytical characterization due to reactivity which necessitates the analysis of both the acid and its potential reaction products. Two of these classes are discussed in detail in the sections below:
Sulfonic Acids
Synthetic Utility
Sulfonic acids are widely used in the pharmaceutical industry to form salts of basic compounds, which modulates the basic compounds physical and/or physiological characteristics.16
Genotoxic Potential
Like other acids, sulfonic acids are susceptible to transesterification reactions in the presence of protic solvents. However, unlike the other acids, the alkyl and aryl sulfonic acids esters (sulfonate esters) have potential genotoxic activity.17-19 For this reason, if at any stage of manufacturing, the sulfonic acid comes in contact with a protic solvent such as methanol, ethanol, or propanol, the corresponding ester must be controlled at the ppm level.
Analysis of Sulfonic Acids
Routine testing of sulfonate esters may not be required if pharmaceutical companies demonstrate that the sulfonate ester is formed below the threshold of toxicological control18,19 but chemical reasoning arguments in the absence of analytical data may not meet the requirements of regulatory agencies. Challenges to developing methods for sulfonate esters include the low sensitivity required and the instability of these compounds in aqueous media.17-19 Because LC-UV typically lacks the sensitivity to quantify genotoxic impurities (GTIs) at low ppm levels, most methods in the literature rely on single ion monitoring (SIM) for GCMS and LCMS,21,22 although HPLC-DAD23 GC-FID14 methods have been reported. Chemical and thermal stability of the sample preparation should be assessed as transesterification occurs more quickly at elevated temperatures and under acidic conditions, while base or water shifts the equilibrium away from the ester.20,18,19 but chemical reasoning arguments in the absence of analytical data may not meet the requirements of regulatory agencies. Challenges to developing methods for sulfonate esters include the low sensitivity required and the instability of these compounds in aqueous media.17-19 Because LC-UV typically lacks the sensitivity to quantify genotoxic impurities (GTIs) at low ppm levels, most methods in the literature rely on single ion monitoring (SIM) for GCMS and LCMS,21,22 although HPLC-DAD23 GC-FID14 methods have been reported. Chemical and thermal stability of the sample preparation should be assessed as transesterification occurs more quickly at elevated temperatures and under acidic conditions, while base or water shifts the equilibrium away from the ester.20
Derivatization of sulfonate esters has been employed to improve the stability of the esters in aqueous media and to improve the sensitivity of the methods utilized in their analysis.24.26 One such derivatization method has shown wide applicability and has been converted into a monograph method in the Eupoean Pharmacopeia. In this method, sodium iodide is reacted with the sulfonate ester in the presence of thiosulfate to form the alkyl iodide, which is readily detected by GCMS. At Genentech, this method was employed for monitoring classic sulfonate esters (e.g. methyl and ethyl methanesulfonate) as well as less widely-used sulfonate esters: the first step of the synthetic route for one development compound utilized methanol to dissolve a starting material edisylate salt; therefore, formation of the monoand di- methyl esters of ethanedisulfonic acid were possible. The EP method was utilized to demonstrate sub-ppm levels of these esters were present in the intermediate formed in Step 1 of the synthesis and in the final API itself.24,27,28
Acyl (Acid) Halides
Synthetic Utility
Acyl chlorides are widely utilized in synthesis due to their ability to form amide bonds with reactive amine groups. Additionally, the Friedel Crafts acylation allows chemists to introduce acyl substituents onto an aromatic ring.29 Acyl chlorides also react with nucleophilic oxygen and nitrogen groups.29
Genotoxic Potential
Due to their high reactivity, acyl halides are alkylating agents and are thus considered genotoxic impurities. However, they are rarely of major concern in final APIs due to their high reactivity, which results in the acyl halide being purged during downstream synthetic steps and/ or reaction work-ups.
Analysis of Acyl Chlorides
From an analytical perspective, acyl chlorides are extremely difficult to characterize (as starting materials or intermediates) or monitor (as potential genotoxic impurities) as they react, often violently, with water, alcohols, and phenols to produce carboxcylic acids or esters and HCl gas. In addition, they sometimes lack stability on silica, the stationary phase of most LC columns.
Normal phase chromatography or SFC may be used to successfully analyze acid chlorides, provided that alcohols and basic modifiers are not used in the mobile phase. This approach is suited to in-process control methods where the disappearance of the reactants and the appearance of the product must be monitored. It is also suitable for the release of raw materials and intermediates. See Figure 1 for a separation of an acid chloride from its corresponding acid hydrolysis product under normal phase conditions.
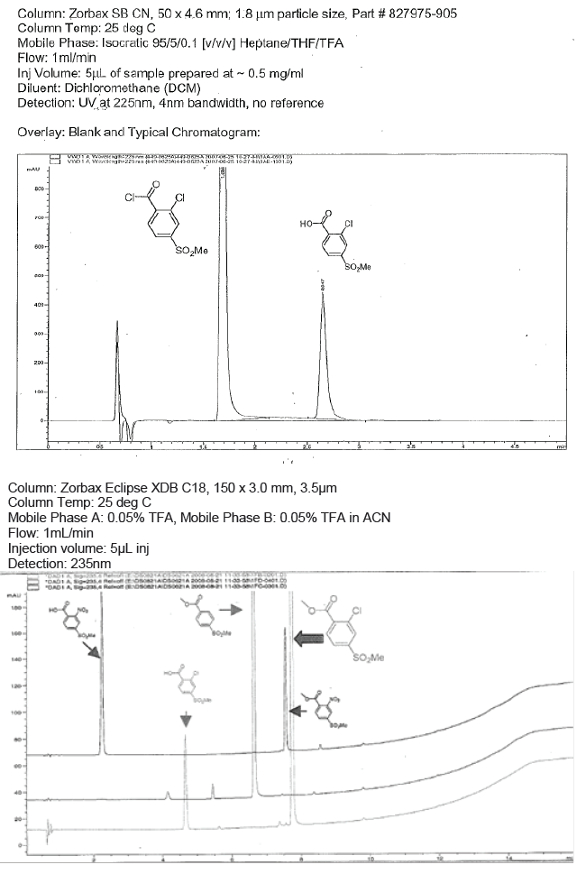 Figure 1. Analysis of Acyl halides via normal phase (top) and derivatization (bottom).
Figure 1. Analysis of Acyl halides via normal phase (top) and derivatization (bottom).If appropriate selectivity cannot be obtained using acid-chloridecompatible mobile phases, or if adequate sensitivity cannot be achieved on an isocratic normal phase method, derivatization followed by reverse phase may be employed.30,31
Due to the reactivity of acyl chlorides, derivatization is widely reported in the literature.32,33 Derivatization of acyl chlorides generates an analyte with adequate stability for analysis by GC for volatile species, or an aqueous-stable analyte amenable to RP-HPLC analysis. If the acyl chloride lacks a chromophore, a functional group with UV absorbance may be incorporated using 4-nitrophenol as a derivatizing agent.34,35 When additional absorbance is not required for detection, simply dissolving the acyl chloride in an alcohol36 quickly converts it to the corresponding ester. The derivatization product should be distinct from any product that could be created during manufacturing and/or stability conditions, eg, methanol should not be used as a derivatizing agent for a compound that contacts methanol during the manufacturing process. When monitoring acyl chlorides as GTIs, coupling derivatization with RP-HPLC and SIM-MS detection is an effective strategy to achieve ppm/ppb level sensitivity. See Figure 1.
Aldehydes
Synthetic Utility
Aldehydes are another class of reactive compounds that are widely used in the synthesis of pharmaceutical APIs. Aldehydes allow chemists to access alcohols via reduction, carboxylic acids via oxidation, and serve as starting materials for nucleophilic addition reactions;37 it’s this flexibility which makes their use so prevalent.
Genotoxic Potential
Aldehydes are known alkylating agents38-40 which can react with and therefore damage DNA.
Analysis of Aldehydes
Like the previous classes of molecules discussed, the reactivity of aldehydes represents an analytical challenge for characterization. Due to the dipole of the aldehyde functional group, the carbon has a partial positive charge41 that is subject to nucleophilic attack by molecules such as water, amines, carbon-based nucleophiles, or even other aldehyde molecules. Aldehydes have a tendency to polymerize, with unsaturated aldehydes having the greatest reactivity. In the presence of oxygen or air, aldehydes oxidize to their corresponding carboxylic acids, with rates depending on the substitution pattern. Aromatic aldehydes are more stable but do oxidize when exposed to air over long periods of time. This process is accelerated with increases in temperature. Basic conditions accelerate oxidation and polymerization, while acidic and basic conditions enhance42 nucleophilic attacks on aldehydes.
The reactivity of the aldehyde (and thus the ability to use standard analytical techniques) must be assessed on a molecule-by-molecule basis. When an aldehyde is sterically hindered or next to electrondonating groups, characterization by RP-HPLC is a viable approach. However, for more reactive aldehydes, alternate analytical techniques are required. Direct inject GC is an ideal methodology for low-molecular weight species because water and other nucleophiles can be avoided. However, care must be taken with diluent selection as the most reactive aldehydes may require non-polar solvents such as hexanes.
For instance, compound X1 degraded rapidly when RP-HPLC was attempted, and was ultimately characterized by GC. Compound X2, however, was successfully analyzed by RP-HPLC. In the case of structure X2, steric hindrance and keto-enol stabilization allowed for successful characterization of X2 and its impurity profile.
In addition to their chemical reactivity, aldehydes are subject to keto-enol tautomerization, and when this reaction is faster than the LC timescale, both species may elute as one broad peak. When the tautomerization is slower than the LC time scale, even fully resolved peaks may show an elevated baseline between them.43 Heating the column may cause the peaks to coalesce.
Amines
Synthetic Utility
Amines are one of the most widely used functional groups in synthetic chemistry, enabling access to a wide array of structures. Common amine reactions include the Schotten-Baumann reaction, C-N coupling, alkylation, acylation, and sulfonation.
Genotoxic Potential
When metabolized, aromatic amines are converted to nitrenium ions that react with the nucleotides of DNA.44-47 Therefore molecules containing this moiety are frequently flagged as GTIs and require trace-level analysis.44,47-51
Analysis of Amines
In general, amines are sufficiently stable for analysis by RP-HPLC. However, historically amines suffered from severe peak tailing in RPHPLC52,53 with the low-pH mobile phases that are desirable due to cleaner baselines afforded by low UV-absorbing additives such as phosphoric and perchloric acids. Traditionally, the lack of retention of amines due to protonation at low pH54 and the presence of peak tailing due to interaction between the protonated amine and free silanols on the silica column required RP-HPLC analysis utilizing high pH mobile phase,55 or ion-pairing reagents, e.g. octanesulfonic acid, that contribute to high baseline absorbance,52,56,57 or chaotropic agents.54,57,58 However, advances in RP-HPLC columns chemistries, including superior end-capping, embedded polar functional groups, and bi- and tri-dentate stationary phase binding, mean that superior peak shapes can now be obtained for amine compounds using RPHPLC and low pH mobile phases.53,55,59 Newer mixed mode reverse phase/cation-exchange stationary phases have been reported to improve the peak shape of basic analytes at low pH,60 and a greater number of stationary phases stable at high pH are now available for use with amine compounds that fail to give good peak shape at low pH even on the best available columns.55,61 As an added benefit, working at a pH well above the amine’s pKa may also improve repeatability of the assay.62
Derivatization of amines is a well-established strategy, especially for highly reactive amines and amines without chromophores. Several well-established derivatization reagents are commercially available, including acylating and silylating agents suitable for primary and secondary amines. More information on derivatization is available in literature.63-66
SFC separations of amines are subject to similar considerations of column technology and mobile phase pH as RP-HPLC separations. However, supercritical CO2 has been shown to react with amine groups to form the corresponding carbamic acid, with primary amines reacting the fastest due to the absence of steric hindrance. Choosing methanol, which preferentially reacts with CO2 to form methylcarbonic acid, as a mobile phase, will protect the amino group of the analyte.67
Aryl amines are relatively unstable (reactive) and are subject to oxidation/ degradation when exposed to air, especially when in solution.68 Salt formation of aryl amine often improves their stability for long-term storage as solids,69,70 and the process chemist may conduct reactions in organic solvents in an oxygen-free environment to avoid degradation. However, exposure to air and water are largely inevitable in the analytical laboratory, making aryl amines a challenge to characterize.
In one extreme case at Genentech, 2-amino-5-fluorobenzene-1,3-diol, a resorcinol compound, was a starting material used in GMP synthesis. It posed a significant analytical challenge for characterization: rapidly degrading, dimerizing, and even trimerizing in solution.71 In addition to its lack of stability, adequate retention could not be achieved on a traditional C18-based column, necessitating the use of a mixed-mode column. A weak cation exchange column gave symmetrical peak shape and adequate retention.
Because the amount of degradants (a/a) increased with the age of the sample preparation, efforts were focused on stabilizing the sample preparation. Multiple diluents (THF, stabilized and unstabilized, ACN, hexane, isopropyl acetate (IPAC)), were screened, with IPAC affording the best solubility and stability. Because solubilizing in IPAC slowed, but did not eliminate degradation, derivitization agents were screened, including methylbenzyl isothocyanate, phenylethyl isothiocyanate, densyl chloride, Fmoc, Boc anhydride, and acetone. These reactions generated multiple side-products and incomplete conversion. Antioxidants such as n-propyl gallate and tocopherol were investigated but failed to slow the degradation. Sparging the IPAC solution with nitrogen or argon also failed to slow the degradation reactions, as did decreasing the concentration of resorcinol in the sample solution. Ultimately, a method with a four-hour solution stability of the IPAC solution was adopted; acceptable as a phase appropriate characterization strategy. See Figure 2.
 Figure 2. Analysis of 2-amino-5-fluorobenzene-1,3-diol.
Figure 2. Analysis of 2-amino-5-fluorobenzene-1,3-diol.Boronate Esters
Synthetic Utility
Boronate acids/esters are widely used in synthetic chemistry in Suzuki coupling, Chan-Lam coupling, Liebeskind-Srogl coupling, conjugate addition, Diels-Alder and C-H functionalization.72,73 Although the boronic acid is the active species in these reactions, the more stable boronate esters are often utilized in a biphasic (organic/aqueous) reaction medium due to their stability under reaction conditions and the ease of characterizing their stoichiometry.74 However, the synthetic advantage of rapid hydrolysis of boronate esters to the reactive boronic acids in situ proves to be a significant challenge for the analytical chemist attempting to analyze boronic esters by RP-HPLC.
Genotoxic Potential
As a class, boronate esters are not known to have genotoxic potential.
Analysis of Boronate Esters
The reactivity of boronic esters with water is pH dependent,75 which necessitates the use of non-aqueous (and typically non-protic) sample diluents below the pKa of the ester. The remainder of this section focuses on stabilizing boronic esters during the analysis itself.
While on-column hydrolysis of boronate esters8 makes them challenging to analyze, several factors that affect the susceptibility of the boronate esters to hydrolysis have been identified. At pH’s greater than the pKa of its corresponding acid, the boronate ester may be stable in aqueous conditions making RP-HPLC analysis feasible.74,76-78 Additionally, electron-donating groups on the aromatic group of a boronate ester can slow hydrolysis by decreasing the Lewis acidity of the boron atom.8 Steric effects also strongly affect the rate of hydrolysis of these esters: greater steric hindrance of the boron atom affords greater resistance to hydrolysis.74,79,80 Regardless of the above stabilizing factors, care should be taken to minimize on-column degradation: modulating the column temperature, the initial gradient composition, and the length of analysis can be utilized to reduce the degree of hydrolysis.81
For boronic esters that are relatively resistant to hydrolysis, low pH RPHPLC separations are possible. Hydrolysis is mitigated in the absence of the silanols that commonly occur in silica-based HPLC columns. In a recent study, selection of an RP-HPLC column with low silanol activity, e.g., the XTerra MS C18, allowed the successful analysis of a variety of boronate esters.8
Alternate approaches are required for boronic esters which are highly susceptible to hydrolysis. The use of a high-pH mobile phase (pH 12) enabled RP-HPLC analysis of such a boronate ester.9 A significant hurdle in this method was the retention of the corresponding boronic acid. Due to its hydrophilic nature, the acid is not retained well at the high pH necessary to stabilize the boronate ester. Polar embedded and mixed mode stationary phases, which increase retention of highly polar analytes, are not compatible with the high pH mobile phase required for this molecule. Instead, an ion-pairing reagent was added to the mobile phase to retain the acid impurity.9 In similar cases, a glucaminium-based ionic liquid82 was used to increase retention of the boronic acid.
Hydrazines/Hydrazones
Hydrazines
Synthetic Utility
Hydrazines represent a class of reactive compounds which are widely used in pharmaceutical synthesis and for which analytical characterization is problematic. This class of compounds is used in the formation of heterocyclic compounds requiring nitrogen-nitrogen linkages [83-87]. They may also be utilized as reducing agents, in WolffKishner reductions [88], and sulfonation reactions.
Genotoxic Potential
Hydrazines are frequently flagged as GTIs. They test positive in the Ames bioassay and are considered potentially carcinogenic in humans, though animal studies showed a significant increase in tumors.89-91
Analysis of Hydrazines
Challenges to the analytical chemists in the analysis of hydrazines include lack of chromophores, lack of retention in reverse phase and gas chromatography, low molecular weight, thermal instability, and the reactivity of the molecules. These factors contribute to poor responses by UV, CAD, FID, and MS. In addition to these challenges, their genotoxic potential requires analysis to the low ppm level. Finally, the analytical chemist should be aware of the potential explosive hazards of this high energy molecule.90,92
The poor response of hydrazine to UV and MS detectors and its lack of retention can be mitigated by derivatization. Hydrazine derivatization methods provide ppm and even ppb levels of detection by improving retention and adding chromophores or MS-ionizable groups,93-96 though hydradzines substituted with larger R groups such as isopropyl or dimethyl may make derivatization hard due to sterics. Many derivatization methods have been developed for use in environmental testing,97-101 and may be applicable to pharmaceutical analysis of substituted hydrazines with modifications to the sample preparation procedure. Analysis of underivatized hydrazine has also been reported in the using alternate retention mechanisms such as ion, ion-exclusion, ion-pair, and HILIC and non-traditional detectors such as CLND and amperometric, conductometric, and potentiometric detectors.102-104
In one case at Genentech, a phenyl hydrazine was used in the synthesis of an early stage project. The R group was non-polar, and adequate retention by HPLC was obtained using a polar-embedded column. The sub-ppm levels desired could not be achieved with UV detection or even MS detection of the parent ion. However, the sensitivity of the method was improved over 10-fold by utilizing MS/MS and monitoring the daughter ion. This technique demonstrated the residual hydrazine to be at adequately low levels in the intermediate formed from the reaction with the hydrazine and was shown to be absent in the final API. Figure 3.
 Figure 3. MS/MS of daughter ion for sub-ppm detection of (2,4-
Figure 3. MS/MS of daughter ion for sub-ppm detection of (2,4-difluorophenyl
)hydrazine.Conclusion
In this review, we have provided a toolkit of analytical techniques and approaches to enable the analysis of reactive molecules used in the synthesis of pharmaceutical products. We have demonstrated that the while derivatization remains a valuable tool for analyzing the most reactive intermediates and starting materials, the analytical chemist can often use the industry-preferred separation and detection by HPLC. Eliminating or reducing components of the mobile phase known to react with the analyte of interest is frequently successful, as is selecting columns with less reactive stationary phases and decreasing overall analysis time. GC remains an important tool, and SFC continues to gain adoption across the industry. By leveraging this range of analytical techniques, the analytical chemist can deliver a high quality, reproducible test methods capable of obtaining repeatable and robust analysis to ensure high quality products and patient safety.
References
- Zhang, T.Y., Process Chemistry: The Science, Business, Logic, and Logistics. Chemical Reviews, 2006. 106(7): p. 2583-2595.
- Webster, A.A., Pharmaceutical Product Stability, in Pharmaceutical Manufacturing Handbook. 2007, John Wiley & Sons, Inc. p. 687-700.
- Li, Y., et al., Limiting degradation of reactive antibody drug conjugate intermediates in HPLC method development. Journal of Pharmaceutical and Biomedical Analysis, 2014. 92(0): p. 114-118.
- ASSESSMENT AND CONTROL OF DNA REACTIVE (MUTAGENIC) IMPURITIES IN PHARMACEUTICALS TO LIMIT POTENTIAL CARCINOGENIC RISK M7 2014.
- Reddy, A.V.B., et al., Identification, control strategies, and analytical approaches for the determination of potential genotoxic impurities in pharmaceuticals: A comprehensive review. Journal of Separation Science, 2015. 38(5): p. 764-779.
- Giraudeau, P. and L. Frydman, Ultrafast 2D NMR: An Emerging Tool in Analytical Spectroscopy. Annual Review of Analytical Chemistry, 2014. 7(1): p. 129-161.
- Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. Cue. 2012: John Wiley & Sons. 768.
- Kumar, A., et al., Accurate Analysis of Boronic Pinacol EstersUsing Low Residual Silanol Silica Based Reversed Phase HPLC. Journal of liquid chromatography & related technologies, 2014. 37(14): p. 1985-1998.
- Zhong, Q., et al., Strategies for the analysis of highly reactive pinacolboronate esters. Journal of Chromatography A, 2012. 1229(0): p. 216-222.
- Orata, F., Derivatization Reactions and Reagents for Gas Chromatography Analysis, in Advanced Gas Chromatography - Progress in Agricultural, Biomedical and Industrial Applications, D.M.A. Mohd, Editor. 2012, InTech.
- Handbook of Derivatives for Chromatography, K. Blau and J.M. Halket, Editors. 1993, John Wiley & Sons.
- Derivatization Reagents For Selective Response and Detection in Complex Matrices, SigmaAldrich, Editor. 2001.
- Analytical Chemistry: An Introduction. 5th ed. 1990: Saunders College Publushing. 642.
- Li, W., Trace analysis of residual methyl methanesulfonate, ethyl methanesulfonate and isopropyl methanesulfonate in pharmaceuticals by capillary gas chromatography with flame ionization detection. J Chromatogr A, 2004. 1046(1-2): p. 297-301.
- Mamilla, Y., et al., A Sensitive and Selective GC–MS Method for Analysis of Process-Related Genotoxic Impurities in Atenolol. Chromatographia, 2010. 71(7-8): p. 733-736.
- Elder, D.P. and D.J. Snodin, Drug substances presented as sulfonic acid salts: overview of utility, safety and regulation. Journal of Pharmacy and Pharmacology, 2009. 61(3): p. 269- 278.
- Elder, D.P., A. Teasdale, and A.M. Lipczynski, Control and analysis of alkyl esters of alkyl and aryl sulfonic acids in novel active pharmaceutical ingredients (APIs). Journal of Pharmaceutical and Biomedical Analysis, 2008. 46(1): p. 1-8.
- GUIDELINE ON THE LIMITS OF GENOTOXIC IMPURITIES. 2006.
- Questions and answers on the ‘Guideline on the limits of genotoxic impurities’. 2010.
- Elder, D.P., et al., The utility of sulfonate salts in drug development. Journal of Pharmaceutical Sciences, 2010. 99(7): p. 2948-2961.
- Raman, N.V.V.S.S., et al., Determination of genotoxic alkyl methane sulfonates and alkyl paratoluene sulfonates in lamivudine using hyphenated techniques. Journal of Pharmaceutical Analysis, 2012. 2(4): p. 314-318.
- Taylor, G.E., M. Gosling, and A. Pearce, Low level determination of p-toluenesulfonate and benzenesulfonate esters in drug substance by high performance liquid chromatography/ mass spectrometry. Journal of Chromatography A, 2006. 1119(1–2): p. 231-237.
- García, A., et al., Development of chromatographic methods for the determination of genotoxic impurities in cloperastine fendizoate. Journal of Pharmaceutical and Biomedical Analysis, 2012. 61(0): p. 230-236.
- Lee, C.R., et al., Determination of polar alkylating agents as thiocyanate/isothiocyanate derivatives by reaction headspace gas chromatography. Analyst, 2003. 128(7): p. 857-863.
- Alzaga, R., et al., A generic approach for the determination of residues of alkylating agents in active pharmaceutical ingredients by in situ derivatization–headspace–gas chromatography–mass spectrometry. Journal of Pharmaceutical and Biomedical Analysis, 2007. 45(3): p. 472-479.
- An, J., et al., A practical derivatization LC/MS approach for determination of trace level alkyl sulfonates and dialkyl sulfates genotoxic impurities in drug substances. Journal of Pharmaceutical and Biomedical Analysis, 2008. 48(3): p. 1006-1010.
- Genotoxic Impurities: Strategies for Identification and Control, ed. A. Teasdale. 2011.
- 2.5.38 Methyl, ethyl, and isopropyl methanesulfonate in active substances, in Euopean Pharmacopoeia Online 8.5. 2015.
- Carey, F.A. and R.J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms. 4th ed. 2000.
- Machado, C.M., et al., Development of an Indirect Reverse Phase Method for the Quality Assessment of an Acyl Halide. Journal of Liquid Chromatography & Related Technologies, 1998. 21(4): p. 575-589.
- Colgan, S.T. and I.S. Krull, Derivatization of Alkyl Halides, Acid Chlorides, and Other Electrophiles with Polymer-Immobilized 8-Amino-2-naphthoxide. Journal of Chromatographic Science, 1988. 26.
- Dahlber, J.A. and I.B. Kihlman, Gas Chromatographic Determination of Chlorinated Acetyl Chlorides and Phosgene Present in Air in Very Low Concentrations. Acta Chemica Scaninaciva, 1970. 24.
- Patai, S., The chemistry of acyl halides. 1972: Interscience.
- Cheh, A.M. and R.E. Carlson, Determination of Protentially Mutagenic and Carcinogenic Electrophiles in Environmental Samples. Analytical Chemistry, 1981. 53: p. 1001-1006.
- Bissinger, J.M., et al., High-performance liquid chromatographic analysis of acid chlorides by pro-derivatization. Journal of Chromatography A, 1983. 268(0): p. 102-106.
- Hasegawa, K., et al., Determination of Acid Chloride by High-Performance Liquid Chromatography. Yakugaku Zasshi, 1981. 101(11): p. 1059-1064.
- Carey, F.A. and R.J. Sundberg, Advanced Organic Chemistry Part A: Structure and Reactions 4th edition. 2000.
- Witz, G., Biological interactions of α,β-unsaturated aldehydes. Free Radical Biology and Medicine, 1989. 7(3): p. 333-349.
- Feron, V.J., et al., Aldehydes: occurrence, carcinogenic potential, mechanism of action and risk assessment. Mutation Research/Genetic Toxicology, 1991. 259(3-4): p. 363-385.
- Snapka, R.M., The SV40 Replicon Model for Analysis of Anticancer Drugs. 1996.
- Klein, D.R., Aldehydes and Ketones, in Organic Chemistry. 2013. p. 7.
- Kohlpaintner, C., et al., Aldehydes, Aliphatic, in Ullmann’s Encyclopedia of Industrial Chemistry. 2000, Wiley-VCH Verlag GmbH & Co. KGaA.
- Moriyasu, M., A. Kato, and Y. Hashimoto, Kinetic studies of fast equilibrium by means of high-performance liquid chromatography. Part 11. Keto-enol tautomerism of some [small beta]-dicarbonyl compounds. Journal of the Chemical Society, Perkin Transactions 2, 1986(4): p. 515-520.
- Benigni, R., et al., Quantitative Structure−Activity Relationships of Mutagenic and Carcinogenic Aromatic Amines. Chemical Reviews, 2000. 100(10): p. 3697-3714.
- Bentzien, J. and I. Muegge, In silico predictions of genotoxicty for aromatic amines Frontiers in Bioscience, 2014. 19: p. 649-661.
- Harding, A.P., et al., Evaluation of aromatic amines with different purities and different solvent vehicles in the Ames test. Regulatory Toxicology and Pharmacology, 2015. 71(2): p. 244-250.
- Shamovsky, I., et al., Theoretical Studies of Chemical Reactivity of Metabolically Activated Forms of Aromatic Amines toward DNA. Chemical Research in Toxicology, 2012. 25(10): p. 2236-2252.
- David, F., et al., Strategic Approaches to the Chromatographic Analysis of Genotoxic Impurities, in Genotoxic Impurities. 2011, John Wiley & Sons, Inc. p. 305-349.
- Vanhoenacker, G., et al., Determination of arylamines and aminopyridines in pharmaceutical products using in-situ derivatization and liquid chromatography-mass spectrometry. Journal of chromatography. A, 2009. 1216(16): p. 3563-3570.
- Liu, D.Q., et al., Analytical control of genotoxic impurities in the pazopanib hydrochloride manufacturing process. Journal of Pharmaceutical and Biomedical Analysis, 2009. 50(2): p. 144-150.
- Snodin, D.J., Genotoxic Impurities: From Structural Alerts to Qualification. Organic Process Research & Development, 2010. 14(4): p. 960-976.
- McCalley, D.V., The challenges of the analysis of basic compounds by high performance liquid chromatography: Some possible approaches for improved separations. Journal of Chromatography A, 2010. 1217(6): p. 858-880.
- Okusa, K., et al., Test compounds for detecting the silanol effect on the elution of ionized amines in reversed-phase LC. Journal of Separation Science, 2010. 33(3): p. 348-358.
- LoBrutto, R., et al., Effect of the eluent pH and acidic modifiers in high-performance liquid chromatography retention of basic analytes. Journal of Chromatography A, 2001. 913(1–2): p. 173-187.
- Kirkland, J.J., M.A. van Straten, and H.A. Claessens, Reversed-phase high-performance liquid chromatography of basic compounds at pH 11 with silica-based column packings. Journal of Chromatography A, 1998. 797(1–2): p. 111-120.
- Köhler, J., et al., Comprehensive characterization of some silica-based stationary phase for high-performance liquid chromatography. Journal of Chromatography A, 1986. 352(0): p. 275-305.
- Dai, J., et al., Effect of anionic additive type on ion pair formation constants of basic pharmaceuticals. Journal of Chromatography A, 2005. 1069(2): p. 225-234.
- LoBrutto, R. and Y.V. Kazakevich, Chaotropic effects in RP-HPLC. Adv Chromatogr, 2006. 44: p. 291-315.
- Freiser, H.H., M.P. Nowlan, and D.L. Gooding, Reversed Phase High-Performance Liquid Chromatography of Basic Drugs on a Silanol Deactivated Support. Journal of Liquid Chromatography, 1989. 12(5): p. 827-843.
- Davies, N.H., M.R. Euerby, and D.V. McCalley, A study of retention and overloading of basic compounds with mixed-mode reversed-phase/cation-exchange columns in high performance liquid chromatography. Journal of Chromatography A, 2007. 1138(1–2): p. 65-72.
- Kirkland, J.J., et al., Stability of silica-based, endcapped columns with pH 7 and 11 mobile phases for reversed-phase high-performance liquid chromatography. Journal of Chromatography A, 1997. 762(1–2): p. 97-112.
- Kirkland, J.J., et al., Bidentate Silane Stationary Phases for Reversed-Phase HighPerformance Liquid Chromatography. Analytical Chemistry, 1998. 70(20): p. 4344-4352.
- Longo, M. and A. Cavallaro, Determination of aromatic amines at trace levels by derivatization with heptafluorobutyric anhydride and gas chromatography-electroncapture negative-ion chemical ionization mass spectrometry. Journal of Chromatography A, 1996. 753(1): p. 91-100.
- Kataoka, H., Derivatization reactions for the determination of amines by gas chromatography and their applications in environmental analysis. Journal of Chromatography A, 1996. 733(1–2): p. 19-34.
- Wu, X., et al., Derivatization of genotoxic nitroaromatic impurities for trace analysis by LCMS. Analytical Methods, 2014. 6(18): p. 7277.
- Hanczko, R., et al., Advances in the o-phthalaldehyde derivatizations. Comeback to the o-phthalaldehyde-ethanethiol reagent. J Chromatogr A, 2007. 1163(1-2): p. 25-42.
- Grand-Guillaume Perrenoud, A., et al., Analysis of basic compounds by supercritical fluid chromatography: Attempts to improve peak shape and maintain mass spectrometry compatibility. Journal of Chromatography A, 2012. 1262(0): p. 205-213.
- Lawrence, S.A., Amines: synthesis, properties and applications. 2004: Cambridge University Press.
- Gould, P.L., Salt selection for basic drugs. International Journal of Pharmaceutics, 1986. 33(1–3): p. 201-217.
- Berge, S., L. Bighley, and D. Monkhouse, Pharmaceutical Salts. Journal of Pharmaceutical Sciences, 1977. 66(1): p. 1-19.
- McMillen, D.F.C., Sou Jen; Nigenda, S. Esther; Malhotra, Ripudaman Retrograde reactions of phenolic coal constituents: self-coupling reactions of dihydroxy aromatic structures. Preprints of Papers - American Chemical Society, Division of Fuel Chemistry, 1985. 30(4).
- Matteson, D.S., Boronic Esters in Stereodirected Synthesis. Tetrahedron, 1989. 45(7): p. 1859-1885.
- Miyaura, N. and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chemical Reviews, 1995. 95(7): p. 2457-2483.
- Hall, D.G., Boronic Acids. 2005.
- Sienkiewicz, P.A. and D.C. Roberts, Chemical affinity systems—I: pH dependence of boronic acid-diol affinity in aqueous solution. Journal of Inorganic and Nuclear Chemistry, 1980. 42(11): p. 1559-1575.
- Babcock, L. and R. Pizer, Dynamics of Boron Acid Complexation Reactions. Formation of 1:1 Boron Acid-Ligand Complexes. Inorganic Chemistry, 1979. 19: p. 56-61.
- Van Duin, M., et al., Studies on borate esters 1 : The ph dependence of the stability of esters of boric acid and borate in aqueous medium as studied by 11B NMR. Tetrahedron, 1984. 40(15): p. 2901-2911.
- Springsteen, G. and B. Wang, A detailed examination of boronic acid–diol complexation. Tetrahedron, 2002. 58(26): p. 5291-5300.
- Bernardini, R., et al., Stability of Boronic Esters to Hydrolysis: A Comparative Study. Chemistry Letters, 2009. 38(7): p. 750-751.
- Gamoh, K. and C.J.W. Brooks, Stability and Reversed-Phase Liquid Chromatographic Studies of Cyclic Boronates. Analytical Sciences, 1993. 9(4): p. 549-552.
- Duran, D., et al., Application of Fast Reversed Phase Liquid Chromatography for Analysis of Pharmaceutical Related Boronic Acid and Boronic Pinacol Ester Functionalized Compounds. Journal of Liquid Chromatography & Related Technologies, 2006. 29(5): p. 661-672.
- Joshi, M.D., et al., Using glucaminium-based ionic liquids for improving the separation of 2-aminopyrimidine-5-ylboronic acid and its pinacol ester by high performance liquid chromatography. Journal of Chromatography A, 2013. 1308(0): p. 161-165.
- Lebedev, O., et al., The use of polyanions of hydrazines in the synthesis of heterocycles. Tetrahedron, 2009. 65(27): p. 5438-5442.
- Corey, E.J. and D. Enders, Applications of N,N-dimethylhydrazones to synthesis. Use in efficient, positionally and stereochemically selective CC bond formation; oxidative hydrolysis to carbonyl compounds. Tetrahedron Letters, 1976. 17(1): p. 3-6.
- Job, A., et al., The SAMP-/RAMP-hydrazone methodology in asymmetric synthesis. Tetrahedron, 2002. 58(12): p. 2253-2329.
- Mäeorg, U. and S. Tšupova, Hydrazines and Azo-Compounds in the Synthesis of Heterocycles Comprising N-N Bond. Heterocycles, 2014. 88(1): p. 129.
- Sharma, S., et al., Review on Synthesis of Bioactive Pyrazoline Derivatives. Chemical Science Transactions, 2014: p. 861-875.
- Kuethe, J.T., et al., A Practical, Kilogram-Scale Implementation of the Wolff−Kishner Reduction. Organic Process Research & Development, 2009. 13(3): p. 576-580.
- Elder, D.P., D. Snodin, and A. Teasdale, Control and analysis of hydrazine, hydrazides and hydrazones—Genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products. Journal of Pharmaceutical and Biomedical Analysis, 2011. 54(5): p. 900-910.
- von Burg, R. and T. Stout, Hydrazine. Journal of Applied Toxicology, 1991. 11(6): p. 447-450.
- Sawatari, K., Y. Nakanishi, and T. Matsushima, Relationships between Chemical Structures and Mutagenicity: A Preliminary Survey for a Database of Mutagenicity Test Results of New Work Place Chemicals. INDUSTRIAL HEALTH, 2001. 39(4): p. 341-345.
- Niemeier, J.K. and D.P. Kjell, Hydrazine and Aqueous Hydrazine Solutions: Evaluating Safety in Chemical Processes. Organic Process Research & Development, 2013. 17: p. 1580-1590.
- Sun, M., L. Bai, and D.Q. Liu, A generic approach for the determination of trace hydrazine in drug substances using in situ derivatization-headspace GC–MS. Journal of Pharmaceutical and Biomedical Analysis, 2009. 49(2): p. 529-533.
- Oh, J.-A., J.-H. Park, and H.-S. Shin, Sensitive determination of hydrazine in water by gas chromatography–mass spectrometry after derivatization with ortho-phthalaldehyde. Analytica Chimica Acta, 2013. 769(0): p. 79-83.
- Smolenkov, A.D., et al., Determination of hydrazine by liquid chromatography with preliminary derivatization with naphthalene-2,3-dialdehyde. Journal of Analytical Chemistry, 2012. 67(4): p. 360-363.
- An, Z., et al., Simultaneous Determination of Hydrazine, Methylhydrazine, and 1,1-Dimethylhydrazine in Rat Plasma by Lc–Ms/Ms. Journal of Liquid Chromatography & Related Technologies, 2014. 37(9): p. 1212-1225.
- Mori, M., et al., Highly sensitive determination of hydrazine ion by ion-exclusion chromatography with ion-exchange enhancement of conductivity detection. Journal of Chromatography A, 2004. 1039(1–2): p. 135-139.
- Davis, I., William E. and Y. Li, Analysis of Hydrazine in Drinking Water by Isotope Dilution Gas Chromatography/Tandem Mass Spectrometry with Derivatization and Liquid−Liquid Extraction. Analytical Chemistry, 208. 80(14): p. 5449-5453.
- Collins, G.E. and S.L. Rose-Pehrsson, Sensitive, fluorescent detection of hydrazine via derivatization with 2,3-naphthalene dicarboxaldehyde. Analytica Chimica Acta, 1993. 284(1): p. 207-215.
- George, M., K.S. Nagaraja, and N. Balasubramanian, Spectrophotometric determination of hydrazine. Talanta, 2008. 75(1): p. 27-31.
- Holtzclaw, J.R., S.L. Rose, and J.R. Wyatt, Simultaneous Determination of Hydrazine, Methylhydrazine, and 1,1-Dimethylhydrazine in Air by Derivatization/Gas Chromatography. Analytical Chemistry, 1984. 56: p. 2952-2956.
- Khan, M., et al., Simultaneous Trace Level Determination of Potentially Genotoxic Hydrazine, Methylhydrazine and Alkylamines in Pharmaceutical Substances by CE Using Indirect Photometric Detection. Chromatographia, 2013. 76(13-14): p. 801-809.
- Smolenkov, A.D. and O.A. Shpigun, Direct liquid chromatographic determination of hydrazines: A review. Talanta, 2012. 102(0): p. 93-100.
- Channon, R.B., et al., Selective Detection of Hydrazine in the Presence of Excess Electrochemically Active Pharmaceutical Ingredients Using Boron Doped Diamond Metal Nanoparticle Functionalised Electrodes. Electroanalysis, 2013. 25(12): p. 2613-2619.