Importance of Spray Dried Dispersions
An increasingly large fraction of drug molecules in development are poorly water soluble (BCS class II and IV) (Butler et al. 2010). A number of solubilized formulation technologies have been developed and applied to achieve adequate oral absorption of such compounds. These technologies include use of complexation (Brewster 2007), lipid-based formulations (Feeney 2016, Kalepu 2013), and solid amorphous dispersions (Friesen 2008, Curatolo 2009, Garunath 2013, Huang 2016), which include materials made by hot melt extrusion (HME) and spray drying. Spray dried dispersions (SDDs), in particular, have found broad use as enabling formulation intermediates for increasing the bioavailability of poorly water soluble drugs (PWSDs).
Encapsulation of Spray Dried Dispersions
Although SDDs have most often been incorporated into immediate release tablets (Wyttenbach and Kuentz 2017), there is also value in the capability to effectively formulate SDDs in hard capsules. Potential advantages associated with encapsulation include the ability to rapidly advance a formulation to the clinic with a ‘powder in capsule’ formulation, extemporaneous preparation for dose-ranging or ‘point’ dosing, formulation blinding, dosage form flexibility to accommodate specific target product profiles (TPPs), and the use of the capsule shell itself as a functional excipient to maintain drug supersaturation upon release in the gastrointestinal (GI) tract. These favorable characteristics of encapsulated SDD formulations have led to efforts to better understand the unique considerations associated with development of an encapsulated presentation of SDDs.
Key Considerations for Encapsulating SDDs
Common challenges associated with encapsulated SDDs include limited drug payload in the unit dosage form and gelation of the hydrated SDD within the capsule, which can lead to poor dispersal of the SDD and reduced dissolution of the drug from the encapsulated formulation. In addition, poor powder flowability of some SDDs (e.g. those with low bulk density) can lead to difficulties in capsule filling. The extent to which powder handling issues arise depends on a number of details, including choice of encapsulation equipment. The following sections describe these aspects of encapsulated SDDs in more detail, and highlight strategies used for formulation and process development.
Drug Loading and Manufacturability
Limited drug payload is often a challenge for encapsulated formulations, including SDDs. Maximum drug loading in the capsule depends upon the mass fraction of drug in the formulation (i.e. SDD plus any excipients), as well as powder density, packing properties, compressibility, capsule size, encapsulator type and process parameters. (Hardy 2003)
Using a gravity fill mechanism, the fill density in the capsule tends to be roughly equal to the measured bulk density of the formulation. SDD particle characteristics, such as bulk density, particle size distribution (PSD) and morphology depend on the polymer and API properties and the spray drying processing parameters. SDD bulk density can vary widely, but is typically in the range of 0.15-0.30 g/cm3 . Figure 1 shows the maximum dose that can be encapsulated in a size 00 capsule (0.91 cm3 volume) as a function of blend density for various drug loadings in the blend. For example, to achieve a 100 mg active dose in a size 00 capsule would require a relatively high active loading of 50% in the formulation, assuming a density of 0.2 - 0.25 g/cm3 , which might be typical of a loose or lightly tamped SDD powder. Increasing the density of the fill and/or drug fraction in the SDD or blend would allow a higher dose in the capsule.
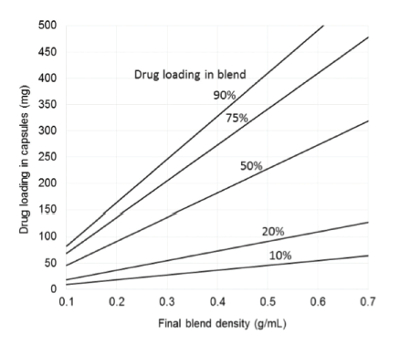 Figure 1. Drug loading in size 00 capsule as a function of blend density for blends of various drug loadings.
Figure 1. Drug loading in size 00 capsule as a function of blend density for blends of various drug loadings.Coating the surfaces of the SDD particles with a small amount of micron or submicron guest glidant particles can increase bulk density and powder flowability for capsule filling (Yang, 2005). The amount of glidant required is typically < 1%wt., and sometimes much less, depending on the relative size and density of glidant and SDD particle. Typical glidants include colloidal silica (e.g. CAB-O-SIL®, AEROSIL®, Syloid®) or magnesium aluminosilicates (e.g. Neusilin®). Coating of SDDs with glidants can typically increase bulk density by 10-80% with a concomitant increase in fill weight using a gravity fill mechanism. Alternatively, drug payload can be increased more significantly by dry granulating the SDD with or without additional excipients to densify particles prior to encapsulation. The increase in density and associated maximum drug loading depends on the composition and initial density of the SDD and any added excipients in the granule. However, a 100-500% increase in density upon dry granulation is typical for SDDs. Despite this more substantial increase compared to coating with glidants, a granulation step adds at least three additional unit operations to the process, diminishing potential process efficiency gains over formulation in tablets.
Capsule filling equipment and mechanism can influence the fill density of powder in capsules. Automatic encapsulation machines based on both dosator and tamping pins compress powder into plugs that are transferred into the empty capsule shells (Jones, 2001). The ability to form plugs and increase fill weight above the measured bulk density during the filling process requires compressibility of the bulk powder, which is a function of PSD, cohesion and mechanical properties (e.g. elasticity) (Sørensen, 2005; Shah, 1986). Formulations containing SDD and excipients tend to demonstrate compressibility under low pressure compression and can give capsule fill densities up to and greater than the measured tapped density of the formulation. However, SDDs coated with glidants without additional excipients tend not to compress much under low pressure tamping, and therefore achieve less gains in density via tamp filling. Manual (e.g. hand-filling) as well as semi-automatic and automatic encapsulation equipment designed to fill powder directly into capsule shells (Hoag, 2017) may be best suited for powder coated SDD in most cases, and are also used widely for formulated powder blends.
Drug Release from Capsules
The target product profile for solid oral immediate release formulations is often rapid and complete dissolution in the stomach and/or upper small intestine to maximize uptake into the intestinal mucosa. Rapid dissolution is facilitated by rapid disintegration and dispersal of the capsule (or tablet) into primary particles. For some encapsulated SDDs, undesired gelling of the SDD can occur within the capsule shell, delaying dispersal of the capsule contents and the associated dissolution of the active ingredient.
The tendency of the SDD to gel varies widely and depends on the particular drug and polymer(s) and their ratio in the SDD. For SDDs that do gel, the phenomenon is related to water ingress into the capsule prior to dissolution of the capsule shell. The largely intact capsule shell keeps the SDD confined during this initial hydration, allowing gelling of the SDD particles (Figure 2) that would otherwise tend to disperse, for example, from an immediate release tablet. The degree of gelation and the impact on dispersal and drug release depend on the properties and loading of the API, the properties of the dispersion polymer, the presence of other excipients, and the particle properties, such as size and density. Gelation also depends upon the opening and dissolution rates of the capsule shell, which are dependent upon capsule shell material, potential interactions between the capsule shell and contents, dissolution media composition and fluid hydrodynamics.
 Figure 2. Physical model of SDD gelling in capsule. Top: rapid water ingress to capsule before capsule shell dissolution leads to SDD wetting and gelling. Bottom: Lower specific surface area of SDD granule, coupled with lower water activity due to rapid osmogen dissolution minimizes gelling of SDD.
Figure 2. Physical model of SDD gelling in capsule. Top: rapid water ingress to capsule before capsule shell dissolution leads to SDD wetting and gelling. Bottom: Lower specific surface area of SDD granule, coupled with lower water activity due to rapid osmogen dissolution minimizes gelling of SDD.In vitro capsule disintegration/rupture times for empty capsules have been shown to be 2-3 minutes for hard gelatin capsules (HGC) versus 4-10 minutes for HPMC capsules in a range of media compositions (Ku et al., 2011). In vivo studies have suggested disintegration times in the range of 6-11 minutes for HGC and 3-13 minutes for HPMC capsules containing lactose mixtures (Tuleu et al., 2007). Complete dissolution of the capsule shell and release of contents takes longer than the opening times. For example, average time for greater than or equal to 90% release of capsule contents was 30, 15, and 15 minutes for compound 1 for HPMC shell 1 (that contains gelling agent), HPMC shell 2 (no gelling agent) and HGC, respectively (Ku et al., 2011). These opening and dissolution times are slow enough even for HGC to promote gelation of the encapsulated SDDs.
Changing the water activity within the hydrating capsule contents through incorporation of an osmogen, such as a salt or sugar, can sometimes mitigate the potential for gelation in such SDD formulations by limiting the rate and extent of water absorption into the SDD. The impact of osmogen addition on the in vitro release of an encapsulated phenytoin SDD, is shown in Figure 3. The effectiveness of the osmogen depends on its water solubility, molar concentration, and method of incorporation within the SDD formulation. The wetting of compacts of SDD blends has, in some cases, shown correlation with in vitro performance of the encapsulated formulations, as shown in Figure 3.
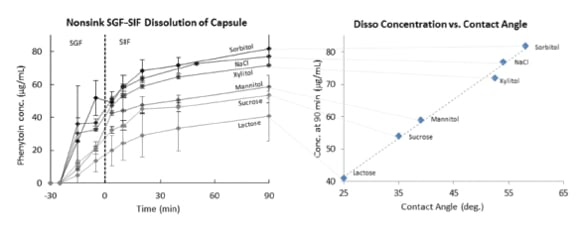 Figure 3. Effect of osmogen on wetting and on dissolution of SDD granules from capsule. For dissolution (left plot) 200 μg/mL phenytoin (SGF, pH 2) to 100 μg/mL (SIF, pH 6.5 in 0.5%wt. SIF powder). Composition 67% SDD granules (25:75 phenytoin:HPMCAS-H) blended with osmogen (31.7%wt.) and Neusilin (2%wt.). Right plot: contact angle of pH 2 HCl solution on a compact of the blend tested in the dissolution test in the left hand plot.
Figure 3. Effect of osmogen on wetting and on dissolution of SDD granules from capsule. For dissolution (left plot) 200 μg/mL phenytoin (SGF, pH 2) to 100 μg/mL (SIF, pH 6.5 in 0.5%wt. SIF powder). Composition 67% SDD granules (25:75 phenytoin:HPMCAS-H) blended with osmogen (31.7%wt.) and Neusilin (2%wt.). Right plot: contact angle of pH 2 HCl solution on a compact of the blend tested in the dissolution test in the left hand plot.Selection of Capsule Shell Material
Traditionally, capsules have been made from gelatin. In recent years, other polymers, such as HPMC, have comprised a larger fraction of the capsule market (Seufert and Vatsa 2016). SDDs can be filled into capsules made from a range of materials. Gelatin dissolves readily at body temperature and has good gelatinizing characteristics favorable for manufacturing, such as gelling, film forming and surface active properties (Ku et al., 2011). HPMC-based capsules have lower water uptake and are less prone to unwanted chemical crosslinking by the capsule contents vs. hard gelatin capsules. Also, HPMC has been shown to sustain supersaturated concentrations of some PWSDs (Gao and Morozowich 2006, Ouellet 2013; Morgen and Goodwin 2016) that are typically formulated as SDDs. As a result, encapsulation of SDDs in HPMCbased capsules can be particularly attractive, as the capsule shell can perform the dual role of both a container for delivering the formulation, and as a functional solubilization excipient. Having the capsule shell effectively solubilize supersaturated drug is particularly valuable for encapsulated SDDs in that it can reduce the need for solubilizing excipients in the fill blend, potentially leaving more space inside the capsule to increase the drug payload. Figure 4 shows in vitro dissolution performance of an SDD comprised of 25:75 erlotinib:poly[(methyl methacrylate)-co(methacrylic acid)] (Eudragit L100), showing improved drug sustainment for the SDD encapsulated in HPMC-based Vcaps® Plus capsules vs. that in HGC.
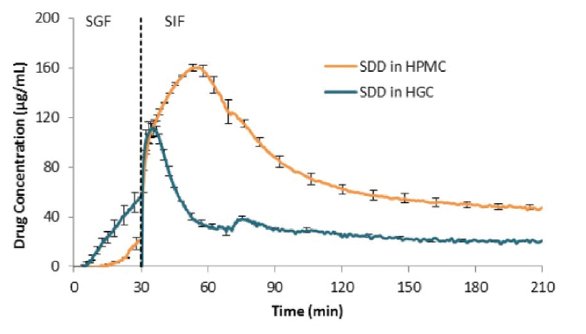 Figure 4. In vitro dissolution profile in a simulated gastric fluid (SGF, 0-30 min) to simulated intestinal fluid (SIF, 30-210 min) transfer test of a 25:75 erlotinib: poly[(methyl methacrylate)-co(methacrylic acid)] (Eudragit® L100) SDD encapsulated in a hard gelatin capsule (HGC) vs. an HPMC (Vcaps® Plus) capsule, showing better sustainment of drug supersaturation for the latter.
Figure 4. In vitro dissolution profile in a simulated gastric fluid (SGF, 0-30 min) to simulated intestinal fluid (SIF, 30-210 min) transfer test of a 25:75 erlotinib: poly[(methyl methacrylate)-co(methacrylic acid)] (Eudragit® L100) SDD encapsulated in a hard gelatin capsule (HGC) vs. an HPMC (Vcaps® Plus) capsule, showing better sustainment of drug supersaturation for the latter.Summary
Spray dried dispersions (SDDs) are increasingly being used to formulate poorly water soluble drugs. The ability to either tablet or encapsulate SDDs offers valuable flexibility in meeting drug development goals, e.g. extemporaneous preparation and blinding for clinical studies, and for meeting various commercial target product profiles. Encapsulated presentations of SDDs require an assessment of maximum unit strength as determined by fill volume, drug loading in the formulation and density of the SDD powder or granulation. In many cases, attention to undesired gelling of the SDD prior to capsule disintegration is necessary to achieve rapid drug release from the capsule. Newer HPMC-based capsules can enhance the performance of SDDs by contributing to sustainment of supersaturated drug levels. Encapsulation of SDDs has been shown to be amenable to a range of encapsulation equipment at various scales, using different fill mechanisms. It is anticipated that the use of both tableted and encapsulated SDD formulations will continue to increase due to the current and future prevalence of PWSDs requiring solubilization technologies.
References
- Butler, J. M.; Dressman, J. B. The developability classification system: Application of biopharmaceutics concepts to formulation development. J. Pharm. Sci. 2010, 99, 4940−4954.
- Brewster, M. E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Delivery Rev. 2007, 59, 645−666.
- Feeney, O.M., Crum, M.F., Claire L. McEvoy, C.L., Trevaskis, N.L., Williams, H.D., Pouton, C.W., Charman W.N., Bergström, C.A.S., Porter. C.J.H.. 50 years of oral lipid-based formulations: Provenance, progress and future perspectives, Adv. Drug Del. Rev. 101 (2016) 167–194.
- Kalepu, S.; Manthina, M.; Padavala, V. Oral lipid-based drug delivery systems − an overview. Acta Pharm. Sin. B 2013, 3, 361−372. Friesen, D. T.; et al. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: An overview. Mol. Pharmaceutics 2008, 5, 1003−1019.
- Curatolo WJ, Nightingale JAS, Herbig SM. Utility of hydroxypropylmethyl cellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu. Pharm Res. 2009;26:1419–31.
- Gurunath, S.; Pradeep Kumar, S.; Basavaraj, N. K.; Patil, P. A. Amorphous solid dispersion method for improving oral bioavailability of poorly water-soluble drugs. J. Pharm. Res. 2013, 6, 476−480.
- Huang, S.; Mao, C.; Williams, R. O.; Yang, C.-Y. Solubility Advantage (and Disadvantage) of Pharmaceutical Amorphous Solid Dispersions. J. Pharm. Sci. 2016, 105, 3549.
- Nicole Wyttenbach a, Martin Kuentz, Glass-forming ability of compounds in marketed amorphous drug products, EJPB (2017), 112 p204-208.
- Seufert, K. and Vatsa, K. HPMC capsules gain credibility as alternative to gelatin, Tablets and Capsules, September 1, 2016.
- Gao, P. and Morozowich, W., Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs, Expert Opin. Drug Deliv. (2006) 3(1):97-110.
- Ouellet D.; Grossmann, K; Limentani, G; Nebot, N.; Lan, K; Knowles, L.; Gordon, M.; Sharma, S.; Infante, J.; Lorusso, P.; Pande, G.; Krachey, E; Blackman, S.; Carson, S. Effects of Particle Size, Food, and Capsule Shell Composition on the Oral Bioavailability of Dabrafenib, a BRAF inhibitor, in Patients with BRAF Mutation-Positive Tumors, J Pharm Sci 102, 3100-3109, 2013.
- Hardy, I., Fitzpatrick, S., Booth, S., “Rational design of powder formulations for tamp filling processes,” J Pharm Pharmacol, 55 (2003) 1593-1599
- Yang, J., Sliva, A., Banerjee, A., Dave, R.N., Pfeffer, R., “Dry particle coating for improving the flowability of cohesive powders,” Powder Tech, 158 (2005) 21-33
- Sørensen, A.H., Sonnergaard, J.M., Hovgaard, L., 2005. Bulk characterization of pharmaceutical powders by low-pressure compression. Pharm. Dev. Tech. 10, 197–209.
- Jones, 2001, The filling of powders into two-piece hard capsules. Int. J. Pharm, 227, 5-26
- Hogan, J., Shue, P., Podczeck, F., Newton, J., “Investigations into the relationship between drug properties, filling, and the release of drugs from hard gelatin capsules using multivariate statistical analysis,” Pharm Res, 13, 6 (1996) 944-949
- Heda, P.K., Muteba, K., Augsburger, L.L., “Comparison of the formulation requirements of dosator and dosing disc automatic capsule filling machines,” AAPS PharmSci, 4, 3 (2002) 1-16.
- Shah, K.B., Augburger, L.L., Marshall, K., “An investigation of some factors influencing plug formation and fill weight in a dosing disk-type of automatic capsule-filling machine,” J. Pharm. Sci., 75, 3 (1986), 291-296
- Hoag, S.W., Capsule Dosage Form: Formulation and Manufacturing Considerations, in Developing Solid Oral Dosage Forms, Pharmaceutical Theory and Practice, 2nd Edition, Academic Press, Elsevier, Inc.
- Ku MS, Lu Q, Li W, Chen Y. Performance qualification of a new hypromellose capsule: Part II. Disintegration and dissolution comparison between two types of hypromellose capsules. Int J Pharm. 2011 Sep 15;416(1):16-24. doi:10.1016/j.ijpharm.2011.02.048. Epub 2011 Feb 26. PubMed PMID: 21356291.
- Tuleu, C., Khela, M.K., Evans, D.F., Jones, B.E., Nagata, S., Basit, A.W., 2007. A scintigraphic investigation of the disintegration behaviour of capsules in fasting subjects: a comparison of hypromellose capsules containing carrageenan as a gelling agent and standard gelatin capsules. Eur. J. Pharm. Sci. 30, 155–251.