Introduction
It is well known that dissolution is an important quality attribute of pharmaceutical dosage forms, not only important during the development phases in order to predict and optimize invivo performance, but also during the commercialization and post-marketing stages of a product. Even for immediate-release (IR) formulations (where the specification for routine quality control testing usually includes Q at a single time-point), reliable information on dissolution profiles is needed to support postapproval changes in accordance to FDA’s SUPAC IR guidance.1 However, the true overall variability of a dissolution method might not be fully understood at the time of regulatory approval given that method reproducibility via an inter-laboratory trial is not required to be thoroughly assessed as part of the marketing application in accordance with ICH Q2.2 It is very common that dissolution testing might be performed routinely at only one laboratory during product development, usually the laboratory that also conducts method validation. But once a product is approved for commercialization, there are multiple drivers for needing to conduct analytical method transfers for dissolution testing including outsourcing of commercial stability activities, onboarding alternate manufacturing facilities and regional regulatory requirements for in-country testing of imported product. This further highlights the importance of understanding the true dissolution behavior (i.e. profile) of a product and the factors impacting the variability of laboratory results.
Solid-oral dosage forms are usually tested utilizing USP Apparatus 1 or 2 as described in USP <711> coupled with UV-end analysis, in many cases requiring HPLC. Unfortunately, dissolution testing using this traditional set-up is prone to variability as reviewed previously3,4 including equipment (e.g. wobble, vibrations, differences in calibration and maintenance practices, etc.) and analyst-toanalyst (e.g. sample introduction, timing, filtering, sampling, etc.) variability. To keep pace with advances in manufacturing utilizing continuous processing and real-time release testing, there is also a desire to find faster and more efficient methods to evaluate dissolution, alternate to sending samples off-line to the quality control laboratory. In terms of compliance, the sources of variability mentioned may provide misleading conclusions depending upon the purpose of the test. For example, it could lead to unnecessary additional testing as part of investigations of perceived trends or false product failures. From the perspective of development, it could lead to an incorrect assignment of criticality when assessing the magnitude of the impact of process parameters and/or material attributes on tablet dissolution.
Given the drivers for developing new approaches in order to understand and reduce variability in addition to improving efficiency, this manuscript focuses on two current approaches to dissolution testing utilized for quality control of commercial solid-oral dosage forms and discusses advantages over traditional approaches as well as challenges in their development and implementation.
The Challenge of Managing a Global Supply Chain
Before discussing technical and efficiency gains, it is important to note that pharmaceutical scientists are faced with the potential constraints of a highly regulated industry and requirements of validation, implementation and approval. The complexity of these requirements is greater when products are marketed globally, and even more for companies that handle low-volume products trying to manage manufactured batches at a global scale. Large volume products could be manufactured for specific markets, making it easier to handle regional method or specification differences, as well as post-approval regulatory changes. For smaller volume products, a company might prefer to split a batch to supply multiple markets, making post-approval change management more challenging and dependent on the rate-limiting regulatory region. Therefore, from a technical perspective, companies often choose to implement newer approaches or technology as an “equivalent” or “alternate” approach to the traditional or “reference” approach. In this manner, regulatory agencies have a side-by-side comparison of the approaches in addition to provide the company flexibility to execute either approach at a multitude of facilities across the globe.
Fiber-Optic Dissolution Testing (FODT)
Since the commercialization of fiber-optic dissolution instrumentation in 1999, several pharmaceutical companies have evaluated the technology for routine applications in a quality control setting given the numerous advantages it offers in terms of speed and minimization of analyst-to-analyst variability. In modern FODT systems, vessels are set up according to USP <711> but are simultaneously and continuously monitored using UV detection with photodiode arrays (PDAs) or charge-coupled device (CCD) cameras; therefore eliminating the need for off-line sampling and/or analysis. In addition, FODT allows the complete understanding of dissolution profiles by high sampling rates, and reproducibly provides much more meaningful data for early time points. The different designs and commercial systems have been evaluated and reviewed by Lu et.al. in terms of hydrodynamics, linear range and light scattering effects.5 Modern instrumentation also offers algorithms to correct for background and/or light scattering effects. Several pharmaceutical applications of the technology have been published, demonstrating that FODT methods can be fully validated in accordance to the requirements of ICH Q2.6,7 However, in most cases, the formulations were selected based on their expected ease of analysis by FODT: single component, high concentration/good response factor and lack of interferences.
The current challenge of FODT still lies in the accurate quantitation of the active component in the presence of interferences, either from excipients or in multicomponent formulations. Several authors have published the use of mathematical models to deconvolute spectral signals in order to quantitate the compound(s) of interest8,9 The advances in FODT instrumentation now allow the acquisition of high volumes of data that can be used for this purpose. For example, the authors have been able to accurately quantitate actives in a commercially available triple combo formulation containing acetaminophen, caffeine and pyrilamine maleate10 utilizing both partial least squares and peak area models. This work included internal development of a custom multivariate analysis tool for more advanced analysis. These three actives possess highly overlapping spectral features, similar release kinetics and a significant difference in concentrations. A formulation like this one would not have been amenable to FODT in the past. A recent MCA software tool is commercially available for two-component formulations.11 This MCA tool uses multiple linear regression and classical least squares analysis and is based on an extinction coefficient calibration matrix derived from a training set built using standard mixtures and is used to calculate individual component concentrations in the unknown mixture. However, interferences and other physical phenomena may cause deviations in Beer’s Law, which might result in the need of highly customized modeling approaches to expand the use of the technology. Without question, there continues to be great interest in expanding the use of FODT to more complex formulations.
In terms of acceptance from a regulatory and compliance perspective, FODT has been deemed as a good alternative to traditional off-line end analysis, in particular for the simpler single-active formulations where equivalency of approaches is easily demonstrated. Internationally accepted guidances for method validation (i.e. ICH Q2) can be easily followed for FODT methods. Gray12 reviewed several aspects to consider from a USP/FDA perspective. It is required that these systems are qualified appropriately for GMP use and are managed through a change management program and appropriate analyst training and standard operating procedures (SOPs). Given the high volume of data collected and analyzed by these systems, all computers need to be compliant with 21 CFR Part 11 and all global data integrity regulations.
However, in the case of multiple component analysis, the analytical method would then rely on a chemometric model which has elevated regulatory and compliance implications. In addition to validating the software tool used for the modeling and calculations, the health of the model itself needs to be evaluated on a periodic basis to ensure that it is still appropriate for the assessment of dissolution in comparison to the reference method. SOPs need to be in place for parallel testing purposes, and all model maintenance or evaluation comparisons against the reference method need to be performed via protocols and documented reports. Updates to the model might require regulatory approval depending on the scope of the change, and different regions might have different reporting categories and timelines for implementation and approval. Bringing it back to the concept of managing a global supply chain, this additional layer of complexity has proved to present challenges with continuous improvement and might ultimately require testing with the reference method while regulatory approvals are on-going.
Spectroscopy-Based Methods – Real Time Release Testing (RTRT)
Continuous manufacturing (CM) platforms have emerged as the ultimate frontier in pharmaceutical drug product manufacturing and has been recognized by FDA and other major regulatory agencies13 as a focal point given all the advantages it can offer, with great potential to improve agility, flexibility and robustness in the manufacturing of pharmaceuticals.14 A continuous process is characterized by the integration of all unit operations and the continuous flow of materials; where input materials are continuously fed into the process while end products are continuously removed.15 A basic building block for a successful continuous operation is extensive development data and process understanding coming as a result of quality-by-design (QbD) principles, leading to a comprehensive control strategy. Therefore, a continuous manufacturing process is a data-rich environment that includes different on-line/in-line measurements and sensing capabilities in order to control set-points for each unit operation, monitor design space parameters and evaluate in-process controls for critical steps. All of this data can then be utilized holistically to evaluate finished drug product quality decisions in real-time, without the need of additional off-line laboratory testing. In other words, real-time release testing (RTRT) is the realization of efficiency gains resulting from the implementation of an advanced control strategy. This includes dissolution evaluation, given the disadvantages and time-consuming nature of the traditional offline testing approach.
In general, both Near-Infrared (NIR) and Raman spectroscopy have been successfully used to model/predict the dissolution behavior of solid-oral dosage forms using multivariate analysis of data, either by partial-least squares (PLS) models or by principal component analysis (PCA) coupled with a regression technique to extract the desired information.16–18 NIR spectroscopy is sensitive to both process parameters (e.g. compaction force) and chemical composition (e.g. water and active content), making this a goto technique for the non-destructive evaluation of tablet quality attributes including dissolution.
There are several options for inputs to dissolution model development given the data rich environment offered by a CM platform. Figure 1 summarizes several approaches possible. When selecting the best approach, beyond technical considerations, quality and regulatory implications are also important from a lifecycle perspective. The first scenario presents a simpler modeling approach, based on measurements and attributes for the tablet formulation only (e.g. using NIR to obtain API concentration in addition to using weight, hardness and thickness measurements from a tablet tester). This approach is simpler, but given the fewer input factors, might be less robust to the additional variation that arises during long-term commercial production. The contrasting third scenario, presents a case of a model potentially built with not only tablet data, but data from both material attributes going into the tablet (e.g. granule particle size) and process parameters (e.g. compression force). In this case, the model would be inherently harder to build and validate, in addition to not allowing room for flexibility in terms of post-approval change management and continuous improvement. However, this modeling approach might be necessary if small changes in process parameters cause a significant impact in product dissolution. The ideal case would be somewhere in between, such as scenario two. In this case, the model is based on material properties for both the tablet in addition to important material (blend or granulation) attributes such as particle size distribution and/or water content. The first step towards building this type of model is to collect reference lab-based dissolution profiles across the design spaces of materials and process parameters. This information is used for the selection of the appropriate dissolution fit model, one which best describes the kinetics and mechanism of the dissolution rates (e.g. Noyes-Whitney, Weibull, etc.). Once the best fit model has been selected, the relationship must be established between the input variables and the ‘rate’ variable in the fit model. Selection of the variables for inclusion into the predictive model should be based upon their statistical significance to the prediction of dissolution or to their known physical impact to dissolution. For example, particle size and bulk density of the APIs, tablet hardness and API concentration would be expected to impact dissolution rate, thus would likely be included in a predictive model. In the event one of those parameters was not statistically significant over the design space or its impact did not exceed a criticality threshold, it likely will not add to the predictive ability of the model. As with any quantitative chemometric model, these relationships should be established across batches, manufacturing design spaces, and lots of in-going materials to ensure accurate predictions across a full range of manufacturing experiences, past and future. The selected parameters serve as the inputs to the quantitative model for the prediction of the ‘rate’ term, which can subsequently be used to calculate the % dissolved at any time point.
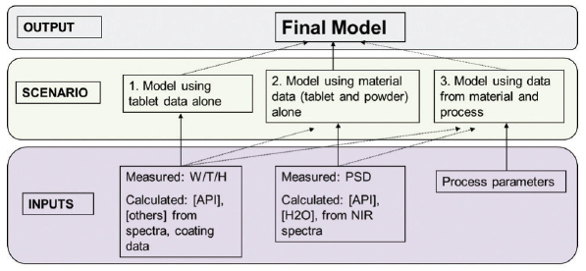 Figure 1. Possible approaches to model development and selection of input parameters.
Figure 1. Possible approaches to model development and selection of input parameters.The described RTRT method offers an alternate / equivalent approach to the reference offline method, and as such needs to comply with applicable guidance documents on method validation and lifecycle approach. Each acquisition and data processing method for the inputs selected is validated appropriately for selectivity, accuracy, precision,linearity and range. In addition, the collective RTRT method is verified against the reference method. During commercial production, the health of all multivariate quantitative and discriminant models is monitored in real-time through multivariate diagnostics (e.g. Hotelling T2 , Q-residuals, Mahalanobis distances) before any predictions are made. If model diagnostics are in trend, the model results in a dissolution-rate prediction which can then be used to calculate a full dissolution profile or the value (i.e. Q) for a specific time-point which can then be compared to the specifications. A summary is presented in Figure 2.
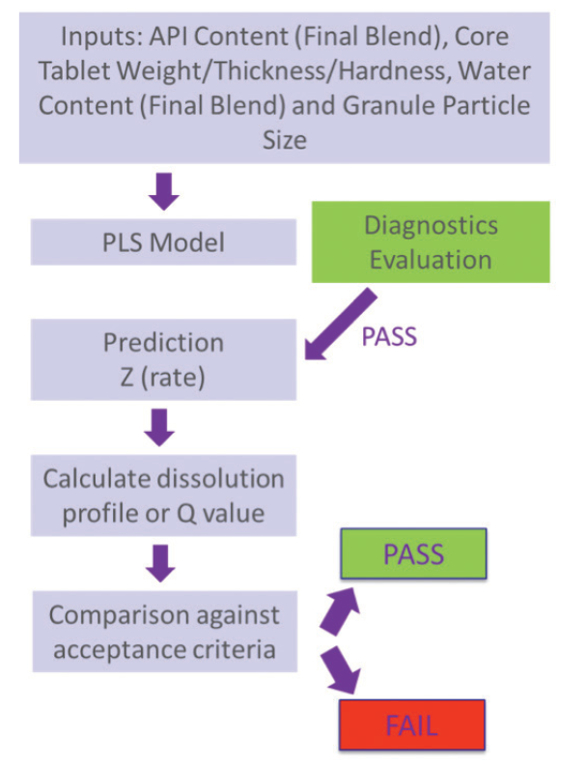 Figure 2. Workflow for model development and use during commercial production.
Figure 2. Workflow for model development and use during commercial production.In accordance with PAT and lifecycle guidelines, the health of the models is also evaluated against the reference method with a predetermined frequency. SOPs for parallel testing and its possible outcomes, including the need for model maintenance, are established as part of the production Quality Systems. Model maintenance and performance trending are some of the challenging aspects of the RTRT approach to dissolution. Model maintenance activities for RTRT methods usually have regulatory impact and the level of impact varies depending on the regulatory region, bringing complexity to supply chain management at the global scale. There are also specific situations where models must be assessed for impact (through change management procedures), including qualification of new materials (e.g. excipient grades/vendors, API manufacturers), process changes (e.g. design space expansions) and non-routine PAT instrument changes as these situations might result in spectral data outside the set used for model development. The performance of these RTRT methods is remarkable in comparison to the off -line reference methods (see Figure 3) and can bring tremendous efficiency gains if adequate procedures for handling model maintenance activities are established, in terms of compliance, regulatory updates and supply chain management.
 Figure 3. Example performance of the RTRT method as compared to HPLC-UV end analysis reference method.
Figure 3. Example performance of the RTRT method as compared to HPLC-UV end analysis reference method.Summary
Dissolution testing continues to be an integral part of the overall pharmaceutical process, used to not only drive formulation development, but quality control and commercial activities required for global supply. Novel dissolution approaches are necessary to match the advances in manufacturing processes as well as to minimize the inherent sources of variability associated with traditional approaches in order to better understand the real contributions from process parameters and material attributes as part of QbD. FODT and RTRT can fulfill this purpose, although careful consideration need to be given to the post-approval lifecycle management of these methods, especially those based on chemometric models.
References
- US FDA Guidance for Industry, Immediate Release Solid Oral Dosage Forms Scale-Up and Post-approval Changes: Chemistry, Manufacturing and Controls, In-Vitro Dissolution Testing and In-Vivo Bioequivalence Documentation, November 1995.
- ICH Q2 (R1), Analytical Methods Validation: Text and Methodology, November 2005.
- Gao Z, Moore TW, Smith AP, Doub, WH, Westenberger BJ. Studies of variability in dissolution testing with USP apparatus 2. J Pharm Sci. 2007, 96(7): 1794-1801.
- Hanson R., Gray V., Handbook of Dissolution Testing, 3rd ed, Dissolution Technologies, 2004.
- Lu X, Lozano R, Shah P. In-situ Dissolution Testing Using Different UV Fiber Optic Probes and Instruments. Dissolution Technologies, Nov (2003), 6 – 15.
- Liu L, Osel T, Hsu J, Greyling J. Evaluation of In-Situ Fiber Optics Dissolution Method for Compound A Extended Release Tablets. Am. Pharm. Rev., Mar (2011).
- Mirza T, Liu Q, Vivilecchia R, Joshi Y. Comprehensive Validation Scheme for In-Situ Fiber Optics Dissolution Method for Pharmaceutical Drug Product Testing. J. Pharm. Sci. 2009, 98(3), 1086 – 1094.
- Wiberg KH, Hultin U-K. Multivariate Chemometric Approach to Fiber-Optic Dissolution Testing. Anal. Chem. 2006, 78(14), 5076 – 5085.
- Nie K, Li L, Li X, Geng D, Zhang Q, Tuo M, Xu P, Chen J. In Situ Fiber-Optic Dissolution Assisted by a Mathematical Separation Model of Dynamic Three-Wavelength K-Ratio Spectrophotometry. Dissolution Technologies, May (2010), 15 – 18.
- Medendorp J, Ryan T, Colón I. Multivariate approaches for the development of quality control in-situ fiber optics dissolution methods for fixed-dose combination tablets. J. Pharm. Biomed. Anal. To Be Submitted.
- Kielt A, Nir I, Seely J, Inman G. Amer. Pharm. Rev., Sep (2016).
- Gray V. Dissolution Testing Using Fiber Optics – A Regulatory Perspective. Dissolution Technologies, Nov (2003), 33 – 36.
- Nasr MM, Krumme M, Matsuda Y, Trout BL, Badman C, Mascia S, Cooney CL, Jensen KD, Florence A, Johnston C, Konstantinov K, Lee SL. Regulatory Perspectives on Continuous Pharmaceutical Manufacturing: Moving from Theory to Practice: September 26-27, 2016, International Symposium on the Continuous Manufacturing of Pharmaceuticals. J. Pharm. Sci. 2017, 106, 3199 – 3206.
- Lee SL, O’Connor TF, Yang X, Cruz CN, Chatterjee S, Madurawe RD, Moore CMV, Yu LX, Woodcock J. Modernizing Pharmaceutical Manufacturing: From Batch to Continuous Production. J. Pharm. Innov. 2015, 10, 191 – 199.
- Fonteyne M, Vercruysse J, De Leersnyder F, Van Snick B, Vervaet C, Remon JP, De Beer T. Process Analytical Technology for Continuous Manufacturing of Solid-Dosage Forms. Trends Anal. Chem. 2015, 67, 159 – 166.
- Hernandez E, Pawar P, Keyvan G, Wang Y, Velez N, Callegari G, Cuitino A, Michniak-Kohn B, Muzzio FJ, Romanach RJ. Prediction of Dissolution Profiles by Non-Destructive Near Infrared Spectroscopy in Tablets Subjected to Different Levels of Strain. J. Pharm. Biomed. Anal. 2016, 117, 568 – 576.
- Navin CV, Tondepu C, Toth R, Lawson LS, Rodriguez JD. Quantitative determinations using portable Raman spectroscopy. J. Pharm. Biomed. Anal. 2017, 136, 156 – 161.
- Pawar P, Wang Y, Keyvan G, Callegari G, Cuitino A, Muzzio F. Enabling real time release testing by NIR prediction of dissolution of tablets made by continuous direct compression (CDC). Int. J. Pharm. 2016, 512, 96 – 107.