Introduction
Quality by Design (QbD), as currently applied to the manufacturing of biological and biotechnological therapeutic products, constitutes a systematic approach to product development which aims at consistently delivering safe and efficacious products of known quality to patients.1,2 The knowledge obtained during development may be used to support the establishment of an operable design space with suitable process controls. These principles are now being considered for application to analytical methods through what is being termed Analytical Quality by Design (AQbD).3 One can find either partial or full implementation of this systematic approach presented in the literature4,5 highlighting the possible benefits of adopting the AQbD approach for the development and implementation of robust analytical procedures.
A vital component of the AQbD approach is to start with the end in mind. This translates to the generation of a predefined analytical target profile (ATP) which explicitly states the intended performance, capability and use of the analytical procedure for ensuring patient safety, manufacturing consistency and efficacy of the product through appropriate monitoring of product critical quality attributes (CQA). The ATP then forms the basis of the generation of the analytical control strategy (ACS).6 The ACS is the sum of steps taken to reduce and/or eliminate risk by ensuring consistent quality of the results generated from the analytical procedure in accordance with the ATP.
Development of an appropriate ACS requires us to understand the following three themes; 1) understanding of fit for purpose, 2) understanding of the method as a process and 3) risk management and control. Understanding fit for purpose assists us in generating an appropriate ATP for monitoring any CQAs that could adversely affect the safety, efficacy and manufacturing consistency of the product. Understanding of the method as a process enables analytical scientists to determine what are the critical method performance factors by determining the relationship between possible variables and their effect on the reportable result of the analytical procedure. Risk management and control is a systemic approach for the assessment of risk to the quality of the reportable value generated from the analytical procedure.
For the purposes of this article the focus will be how to utilize design of experiments (DoE) principles for developing robust analytical methods for quality control (QC) environments by initially understanding the method as a process and secondly by demonstrating robustness.
How Design of Experiments (DoE) Principles Can Be Utilized in Developing Robust Analytical Methods for QC Environments
In current practice, analytical procedures are typically developed utilizing a one factor at a time approach (OFAT), in which one parameter is optimized in isolation with the others remaining constant. By following this approach for method development no information is obtained around how method parameters interact with each other and therefore impact the results obtained. This can lead to analytical procedures with very narrow robust ranges of operation. Hence this strategy has the potential for an increase in risk of method failure after the analytical procedure is transferred from the development environment into an internal or external QC laboratory.
The Design of Experiments (DoE) methodology is a test or series of tests in which purposeful changes are made to the input variables (factors) of a process so that we may observe and identify corresponding changes in the output response. In short, DoE is an approach that can be used to determine cause and effect relationships in a stepwise manner (Figure 1). In the context of developing robust methods for the QC environment, DoE can be utilized in a variety of ways. For example; determining, identifying and evaluating the effect of the most influential method parameters and any interactions between method parameters within the design space so that the method can be developed and then validated in accordance to the guidance provided by the International Conference on Harmonization (ICH) quality guideline Q2: Validation of Analytical Procedures.7 Or secondly, for the purposes of demonstrating robustness of the analytical procedure by assessing the capacity of the method to remain unaffected by small variations in the method parameters, so that it can perform to the requirements stated in the ATP and can be successfully transferred to QC laboratories with a reduction in the risk involved.
 Figure 1. The stepwise manner in which the design of experiments (DoE) methodology can be utilized to develop and implement robust analytical procedures in a quality control (QC) laboratory environment.
Figure 1. The stepwise manner in which the design of experiments (DoE) methodology can be utilized to develop and implement robust analytical procedures in a quality control (QC) laboratory environment.One challenge to analytical development is that the timelines can often be compressed for various reasons. For novel molecules, the sooner methods are optimized, the lower the risk will be to overall product development. By utilizing the DoE methodology for method development, optimization and assessing robustness we can increase our scientific understanding of the method in the same and often shorter time frames than if the OFAT approach is used. Therefore, reducing the risk associated with the results obtained from the analytical procedure in a highly cost-effective manner.
Herein I will describe a case study for how to use DoE principles to optimize the analytical unit operation of sample preparation for a platform analytical procedure for novel biopharmaceutical molecular formats.
The use of platform analytical methods for common product types such as, monoclonal antibodies (mAbs) has reduced the uncertainty/ risk for the manufacturing organization leading to less resource being required for method development, qualification and eventually validation. With the industry producing products with an everincreasing complexity e.g. fusion proteins, antibody drug conjugates and bispecifics to patients in the form of new medicines the use of platform methods becomes an increasingly challenging approach to continue with. Kovacs et al.6 presented that an analytical method can be thought of as three analytical unit operations; sample preparation, measurement and replicate strategy. For platform methods, the measurement and the replicate strategy are defined in the standard operating procedure (SOP) leaving only the product specific analytical unit operations to be investigated and in this situation, this leaves only the sample preparation as the analytical unit operation requiring optimization for analysis of these novel molecular formats. The sample preparation information can be captured in an associated technical document to the SOP for the analyst to follow in the laboratory.
After generation of the ATP, where we have defined the requirements of the analytical procedure the first step towards developing a robust analytical procedure is to screen for the initial method conditions and to determine what are the main factors that affect the method performance, followed by optimizing these factors and then demonstrating robustness of the final conditions. A Placket Burman design is an economical screening design and is an appropriate approach for screening multiple factors simultaneously. For this initial step of the process it is important to choose appropriate ranges for the factors under assessment. Typically, the ranges selected should be wide enough to see an effect but not so wide so as to not be relevant to the method. Characteristically, the ranges should be two to three times the level of process control. For example, if pH is to be controlled to 7.4 ± 0.2 then we would examine a range from 7.0 to 7.8 as part of the screening DoE. By using a screening design such as a Plackett Burman model we will not gather any information if there are any secondary interactions between factors. However, we will be able to identify the main factors that significantly affect the results obtained and this can allow us to remove any factors from future designs that are shown not to be significant for the reportable result obtained from the procedure.
Once the main factors have been identified the next step is to optimize these factors so that we can meet the method performance criteria started in the predefined ATP and then define our method operable design region (MODR). A fractional factorial model is an appropriate design to provide specific combinations of the factors under investigation over the ranges to efficiently test their impact as main effects and the interactions between these factors and is therefore very well suited to factor optimization. A fractional factorial design consists of the factors under investigation at two levels (-1, +1) and with at least one center point, so as to be able to determine if there is curvature in the model. It is important to assess the model fit for the data obtained from this optimization step is good as this will provide confidence that there is predictive power within the design space for between the actual and predicted results (Figure 2). A poor model fit does not afford us this predictive power therefore, additional levels will need to be added to the design to improve the model fit through augmentation. Additionally, if curvature is observed then additional experimentation would be required to be able to accurately model the curvature present and then give us confidence in the predictive power of the model determined for the reportable results obtained. Once a good model fit is obtained and we have sufficient confidence in the predictive power of the model for the design space under investigation we can then start to determine our method operable design region (MODR) (Figure 3) for the critical method attributes through robustness studies and then define our final analytical procedure conditions noted in the SOP or technical document.
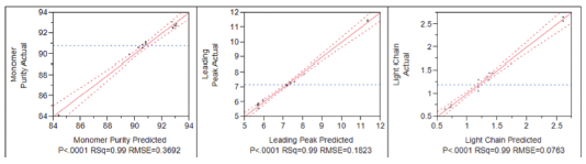 Figure 2. Results from the fractional factorial model design used for method optimization. The strong linear correlation observed between the predicted and actual data indicates the model has a strong predictive power.
Figure 2. Results from the fractional factorial model design used for method optimization. The strong linear correlation observed between the predicted and actual data indicates the model has a strong predictive power. Figure 3. Contour maps from the DoE for study for sample preparation optimization. The robust method operable design region (MODR) is highlighted by the red squares.
Figure 3. Contour maps from the DoE for study for sample preparation optimization. The robust method operable design region (MODR) is highlighted by the red squares.Prior to authoring, review and approval of the SOP with the new analytical procedure information it is very important to perform a robustness assessment of the final optimized conditions to reduce the risk of possible future analytical procedure failures. As mentioned previously, for the optimization step of the method development process the ranges selected were characteristically, the ranges should be two to three times the level of process control. For assessing the robustness of the analytical procedure, the ranges of the factors under investigation should now be tightened to be representative of the level of acceptable process control. For example, if the factor under investigation is temperature then for the optimization DoE the range for this factor could be 65°C ± 5°C and where if 65°C is the determined optimum, for the robustness DoE the range could be 65°C ± 2°C. The aim of the robustness DoE is that for all conditions investigated the reportable results obtained are within the acceptable level of variation stated in the ATP and that additionally all criteria of the ATP be met. To mitigate the risk further the final analytical procedure conditions can be performed by additional scientists on different instrumentation to gain an additional understanding of the performance of the analytical procedure before implementation.
Summary
A DoE approach for how to optimize and demonstrate robustness of the analytical unit operation for sample preparation of novel molecular formats for a platform analytical procedure has been presented in this article. This sequential process built on objective data and not personal intuition guides the decision making and affords the mitigation of risk in comparison to utilizing a OFAT methodology and additionally allows for investigating factor interactions which is also not possible from the OFAT approach leading to the generation of more robust analytical procedures suitable for the QC environment. A supplementary benefit of utilizing DoE software is that graphical data representation allows for easy analysis of the reportable results obtained and that the large amount of data generated from utilizing DoE methodologies can be an important aspect of an AQbD approach with regards implementing an appropriate ACS for the analytical procedure.
Acknowledgements
The author would like to acknowledge the following people for their on-going support and assistance Guillermo Miro-Quesada, Christopher Larkin, Douglas Johnson, Kristin Schultz-Kuszak, Derek Schildt, Eric Meinke and Xiangyang Wang
References
- ICH, Q8(R2): Pharmaceutical Development (ICH, August 2009)
- ICH, Q11: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) (ICH, May 2012)
- P. Borman, P. Nethercote, M. Chatfield, D. Thompson, K. Truman, The application of quality by design to analytical methods, Pharm. Tech. 31 (2007) 142–152
- J. Kochling, W. Wu, Y. Hua, Q. Guan, J. Castaneda-Merced, A platform analytical quality by design (AQbD) approach for multiple UHPLC-UV and UHPLC-MS methods development for protein analysis, Journal of Pharmaceutical and Biomedical Analysis. 125 (2016) 130–139
- Y. Li, G.J. Terfloth, A.S. Kord, A systematic approach to RP-HPLC method development in a pharmaceutical QbD environment, Am. Pharm. Rev. 12 (2009) 87–95.
- E. Kovacs, J. Ermer, P. L. McGregor, P. Nethercote, R. LoBrutto, G. P. Martin, H. Pappa, Stimuli to the Revision Process: Analytical Control Strategy, 42 (5) (2016)
- ICH, Q2(R1): Validation of Analytical Procedures: Text and Methodology (ICH, November 2005).