It has been approximately eight years since the Biologics Price Competition and Innovation Act (BPCIA) was signed into law by President Obama as part of healthcare reform. Among other things, the BPCIA created an abbreviated pathway for regulatory approval of therapeutic biologics. The rationale behind creating this new pathway was to encourage competition among biologics manufacturers to reduce prices and increase patient access to biologic therapeutics.
Similar to the Drug Price Competition and Patent Term Restoration act of the 1980s (informally known as the “Hatch-Waxman Act”), the BPCIA permits biosimilar applicants to rely on safety and efficacy data from prior studies conducted with the approved (reference) product. While there are a few similarities between the provisions of the BPCIA and the Hatch-Waxman Act, the complexity of biologics necessitated new guidelines for demonstrating safety and efficacy for biosimilar products. Unlike small molecule pharmaceuticals that can be copied with precision, biologics are subject to alterations in structure, function, and even chemical composition that can result from small changes in the manufacturing process or environment. These alterations make pharmacy-based substitution of a biosimilar product for a referenced product substantially more complex.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
Despite an uptick in the number of abbreviated biologic license application (aBLA) approvals, five in 2017 alone, only three biosimilar products have entered the market. The first entrant launched in the fall of 2015 as Zarixo (filgrastim-sndz), Sandoz’s biosimilar to Amgen’s Neupogen. Following five quarters of sales, Zarixo accounted for approximately 17% of sales in the short-acting filgrastim market.1,2 In comparison, small molecule branded products facing generic competition for the first time in 2011-2012 saw market shares erode roughly 80% within six months after entry of the first generic.3
What is behind this seemingly slow rate of biosimilars market entry and subsequent growth in market share? Several factors may be contributing including regulatory uncertainties, slow adoption by physicians and payers, and extensive patent litigation post-approval.
Regulatory Hurdles
Even though the BPCIA created an abbreviated pathway for approval of biosimilars in 2010, it took the Food and Drug Administration (FDA) another two years to issue the first draft guidance for developing and registering a biosimilar and three additional years until the FDA approved the first biosimilar.
The agency established two classes of biologic drugs that can be licensed under the BPCIA: those that are “biosimilar” and those that are “interchangeable”. To be approved as a “biosimilar," an abbreviated biologic license application (aBLA) must contain pre-clinical and clinical data sufficient to demonstrate that the biosimilar product is “highly similar” to and without “clinically meaningful differences” from the approved reference product.4 The applicant must provide information about its manufacturing process and facility to assure the biological product will have comparable safety, purity and potency to the reference product. The applicant must also demonstrate the condition or conditions of use prescribed, recommended, or suggested in the proposed labeling were previously approved for the reference product, and the route of administration, dosage form, and strength of the biosimilar product must be the same as the reference product.
For a biosimilar applicant to receive approval of a biosimilar as an interchangeable drug, the applicant must demonstrate that the biosimilar product is sufficiently similar to the reference product and is expected to produce the same clinical result as the reference product in any given patient.5 For any biosimilar product that is to be administered more than once to the same individual, the applicant must also show that alternating or switching between the reference and biosimilar product has the same risk in terms of safety and efficacy as not alternating or switching products.
None of the nine approved biosimilars has interchangeability status. While some perceive that interchangeability will dramatically affect the biosimilars market share, others are more skeptical whether the additional investment will provide the biosimilar applicant adequate financial return.
To help address this concern, the BPCIA affords the first licensed interchangeable product a period of market exclusivity as to other interchangeable biological products for at least one year. Subsequent interchangeable biosimilar applicants cannot be approved for any condition of use until the earliest of the following occurrences:
- One year after the first commercial marketing of the first interchangeable product;
- 18 months after a final court decision in favor of the applicant with respect to all patents in suit or a dismissal of the complaint with or without prejudice;
- 42 months after approval of the fi rst interchangeable product if litigation is ongoing; or
- 18 months after approval of the first interchangeable product if the first interchangeable applicant has not been sued.6
This exclusivity does not, however, prevent approval of “biosimilar,” i.e., not interchangeable, products for the same reference product.
Some perceive that interchangeability, i.e. allowing pharmacy substitution of a biosimilar product, will greatly increase the uptake of biosimilars in the market while others disagree. Unlike small molecule therapeutics that are primarily prescribed for self-administration by the patient, many of the biologic therapies are IV infusions administered in a medical facility under the direct supervision of a physician. Thus, pharmacy substitution may not be particularly relevant. In addition, it is still not clear whether approval of a biosimilar as “interchangeable” for a single indication will automatically afford the biosimilar approval for all indications approved for the reference product, or if the biosimilar applicant will still have to provide sufficient data pertaining to each indication. Furthermore, biosimilar applicants will have to determine whether the investment in additional studies to achieve interchangeable status will provide a better return than investing in patient and physician education and marketing of noninterchangeable biologics.
The FDA continues to introduce initiatives to help speed the approval of biosimilars. The agency has issued multiple guidance documents pertaining to demonstrating biosimilarity to a reference product, including scientific, clinical and quality considerations, demonstrating interchangeability, and most recently statistical approaches for evaluating analytical similarity.7 In its 2018 Strategic Plan, the agency indicates it will take new steps to make the process for developing and approving biosimilar drugs more efficient. The FDA will advance these new policies through its Biosimilar Innovation Plan (BIP) forthcoming sometime in 2018. In addition, President Trump signed a bill that reauthorized the FDA user fee programs for 2018-2022, allowing the agency to hire more staff to provide additional communications between the agency review teams and biosimilar applicants.8
Legal Hurdles
Despite the patent litigation framework provided in the BPCIA, a lack of judicial precedent put early biosimilar applicants at a disadvantage. With the first patent case fi led by Amgen against Sandoz in 2014, much of the early litigation centered on interpretation of certain provisions in the BPCIA.
Following the submission and acceptance of an applicant’s aBLA, the BPCIA provides that the applicant “shall” supply the reference product sponsor (RPS) a copy of the aBLA and associated manufacturing information to allow the RPS to prepare a list of patents that the RPS identifies as potentially infringed by the proposed biosimilar.9 This initial list triggers subsequent rounds of disclosures by both sides, purportedly to result in an agreed upon list of patents that the RPS and the applicant agree must either be litigated or licensed. In addition, the applicant must notify the RPS at least 180 days in advance of marketing its approved biosimilar.10
As the first patent matter played out between Amgen and Sandoz, Sandoz declined to provide a complete copy of its aBLA or the manufacturing information related to its proposed biosimilar. Instead, Sandoz adopted an interpretation of the disclosure provisions that despite the seemingly compulsory “shall” language, the disclosure provisions were not mandatory and not enforceable by injunction.
The Supreme Court ultimately agreed with Sandoz, reasoning that the BPCIA provides a remedy for the RPS ((i.e. the right to bring an immediate infringement action) if the applicant does not produce the aBLA and manufacturing information and, therefore, the Court should not provide additional remedies.11 The Supreme Court also weighed in on interpreting a second provision of the BPCIA that pertains to the 180-day notice of the first commercial marketing. Shortly after Sandoz received notice that the FDA accepted its aBLA for review, Sandoz notified Amgen of its intent to market its biosimilar immediately after FDA approval. Following approval, Sandoz again notified Amgen of its commercial marketing. Amgen objected to Sandoz’s interpretation of the 180-day notice period, alleging that Sandoz’s notice was premature and that the 180-day notice of commercial marketing is valid only after the FDA has licensed the biosimilar product. Although the Federal Circuit agreed with Amgen’s interpretation, the Supreme Court sided with Sandoz, finding that the applicant may provide notice of commercial marketing either before or after the FDA approves its biosimilar product.12
Despite the clarity provided on two key provisions of the BPCIA, many questions remain. For example, what are the available remedies for RPS and applicants alike, including the scope of patents that can be the subject of a declaratory judgment action if the applicant opts out of the disclosure requirement? The BPCIA provides that the RPS can bring a declaratory judgment action regarding “any patent that claims the biological product or use of the biological product”.13 The BPCIA does not specify if the RPS can also bring a declaratory judgment action for any process patents. In addition, is the RPS limited to reasonable royalty damages if the parties engage in the patent list exchange and a suit is brought more than 30 days after the information exchange? These and other questions are under judicial review and more clarity is expected in the coming year.
A number of infringement actions will see substantial activity in 2018. At least seven biologics were subject to or are the current subject of ongoing litigation including Remicade, Neupogen, Epogen, Enbrel, Humira, Avastin, and Herceptin. Many of the pending litigations involve multiple patents. For example, Genentech recently brought a suit against Pfizer for infringement of 40 patents based on its trastuzumab aBLA (biosimilar to Herceptin).
Physician, Patient and Payer Acceptance
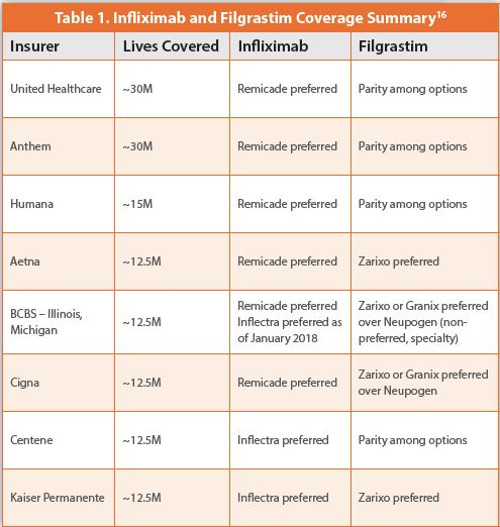
One estimate of the potential savings from the introduction of biosimilars is a reduction of approximately $54 billion in direct spending from 2017 to 2026, with a range of $24 to $150 billion.14 However, a significant amount of that savings depends on insurers, prescribers, and patients accepting regular use of biosimilars in healthcare. With only three approved biosimilars for two reference products on the market, it is difficult to determine the overall acceptance of biosimilars. However, early data suggest that payers have not overwhelmingly adopted biosimilar products as the preferred choice for treatment. In fact, payers may be persuaded to stick with an innovator’s biologic unless the biosimilar provides discounts of 40-60% off net price.15
Acceptance of biosimilars by physicians has also lagged. In a recent survey of specialists and primary care physicians by the online physician platform and community QuantiaMD,17 78% of doctors polled said they were familiar with the term “biosimilar,” but only 17% of prescribing specialists felt very likely to prescribe them. In addition, only 12% of the prescribing specialists were “very confident” that the biosimilars are as safe as the original version of the drug and 80% said they would need to learn more about biosimilars.18
Conclusion
Although the penetration of biosimilars into the biologics market is smaller and slower than some may have anticipated, innovators and biosimilar applicants alike will continue to face many challenges including legal, regulatory, payment and pricing challenges. Although the BPCIA is closing in on its eighth year, it appears that it will be at least another eight years before the true effects of the BPCIA on competition and cost savings can be more accurately assessed.
Author Biography
With over two decades of experience in the pharmaceutical and biotechnology industries, Jennifer Fox has built a solid reputation as a trusted advisor to biotechnology and pharmaceutical corporations locally, nationally, and worldwide. She has the ability to forge strong relationships with her clients and strategically counsel them on key intellectual property issues. Her practice includes patent portfolio procurement and management, licensing and other agreements, IP due diligence and transactional support, and post-grant proceedings.
References
- In addition to Neupogen, Teva’s filgrastim product Granix launched in late 2013 and accounted for approximately 20% of sales ending 4Q 2016. Mulcahy AW, Hlavka JP, Case SR. Biosimilar Cost Savings in the United States: Initial Experience and Future Potential. Santa Monica, CA: RAND Corporation; 2017. Available at: https://tinyurl.com/y7re6ecn, Accessed April 1, 2018
- Mulcahy AW, Hlavka JP, Case SR. Biosimilar Cost Savings in the United States: Initial Experience and Future Potential. Santa Monica, CA: RAND Corporation; 2017. Available at: https://tinyurl.com/y7re6ecn, Accessed April 1, 2018
- Grabowski H, Long G, Mortimer R. Recent trends in brand-name and generic competition. J. Med Econ 2013; 1-8.
- 42 U.S.C. 262(i)(2)
- 42 U.S.C. 262(i)(3)
- 42 U.S.C. 262(k)(6)
- https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm290967.htm, Accessed April 1, 2018
- https://www.fda.gov/ForIndustry/UserFees/BiosimilarUserFeeActBsUFA/default.htm, Accessed April 1, 2018
- 42 U.S.C. 262(l)(2)
- 42 U.S.C. 262(I)(8)(A)
- Amgen v. Sandoz, 137 S. Ct. 1664 (2017)
- Amgen v. Sandoz, 137 S. Ct. 1664 (2017)
- 262(l)(9)(C)
- Mulcahy AW, Hlavka JP, Case SR. Biosimilar Cost Savings in the United States: Initial Experience and Future Potential. Santa Monica, CA: RAND Corporation; 2017. Available at: https://tinyurl.com/y7re6ecn, Accessed April 1, 2018
- Stanton D. Remicade Biosimilar: J&Js “Fear and Loathing” Subdued as Pfizer Slugs it Out. Biopharma Reporter (website). September 14, 2017. Available at: https://tinyurl.com/yaeq3mq2, Accessed April 1, 2018
- Adapted from Hoffmann M and Barry J. US Biosimilars 2018: Opportunities and Challenges. Back Bay Life Sciences Advisors Whitepaper, Jan. 2018. Available at https://bblsa.com/whitepaper-us-biosimilars-2018-opportunities-and-challenges/, Accessed April 1, 2018
- https://www.quantiamd.com/, Accessed April 1, 2018
- Kuonen S. Success of Biosimilars Depends on Physician Education. Navigate Perspectives: Blog. http://www.navigatecorp.com/success-of-biosimilars-depends-on-physician-education/, Accessed April 1, 2018