Abstract
Kolliphor® HS 15 is an exceptional solubilizer with attributes suited for parenteral and oral formulation of poorly soluble molecules. As a consequence, it is primarily used in screening of new chemical entities (NCEs) and also used as a vehicle in delivery of molecules encapsulated in micelles or micro-emulsions. Its physicochemical and application relevant characteristics are attributed to a unique structure comprised of 12-hydroxystearic acid (lipophilic moiety) and polyethylene glycol (hydrophilic moiety). As a solubilizer with exceptional safety and toxicity profiles, and regulatory perspectives, it offers unique advantages over many other solubilizers of its class for injectable (IV, infusion, im, sc) formulations, in self-emulsifying drug delivery systems (SEDDS/SMEDDS) and lipid based nanoparticulates.
Keywords: Kolliphor® HS15, Solutol® HS 15, polyoxyl 15 hydroxystearate, macrogol 660 hydroxystearate, solubilizer, self-emulsifying systems; SEDDS and SMEDDS, critical micelle concentration, lipid nanocapsules, parenteral excipient, oral drug delivery, histamine release, Pgp inhibition, leachable and extractable.
Introduction
Kolliphor® HS 15 (formerly regarded as Solutol® HS 15) a non-ionic surfactant, has been used as a solubilizer in parenteral and oral formulations for over several decades.1 Solubilizers like Kolliphor® HS 15 and others have been a subject of continued interest in the industry since the number of poorly soluble new chemical entities (NCEs) is constantly increasing. Thus, the effort continues to bring these molecules to pipeline for development in oral and parenteral formulations.2 Kolliphor® HS 15 outperforms in its class of many lipid based solubilizers, and hence, is frequently used in in vitro screening and in vivo efficacy studies of compounds.3,4 Kolliphor® HS 15 bears the hallmarks of an exceptional solubilizer with inherent amphiphilicity stemming from its combined lipophilic and hydrophilic characteristics derived by reacting 1 mole of 12-hydroxystearic acid with 15 moles of ethylene oxide. It has the hydrophilic lipophilic balance (HLB) of 16 and a critical micelle concentration (CMC) of 0.005%-0.02%. Due to its unique solubilization characteristics and safety profiles, this excipient has also been used in ophthalmic, suppository, macromolecule and protein formulations.5,6
Kolliphor® HS 15, a monographed excipient comprised of about 65%-70% of ethoxylated mono- and di-ester components, where both the carboxylic group as well as the hydroxy group can be ethoxylated, and about 30%-35% of free PEGs, has a unique composition that does not only act as a solubilizer but also as a co-solvent for poorly soluble molecules.7 In addition, it offers advantages over highly polymeric solubilizers because it spontaneously self-assembles into micellar structures consisting of a large number of molecules. Kolliphor® HS 15, a non-ionic compound in nature, is pH independent and forms micelles typically ranging 10 – 15 nm. It enables an efficient drug loading into its hydrophobic interiors and generates a thermodynamically stable, highly concentrated system, a pre-requisite for longer systemic circulation, faster absorption and enhancement of bioavailability.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
Approved by FDA in injectable and ophthalmic drugs, and by other regulatory agencies globally in many other drug products for humans and animals, Kolliphor® HS 15 has created an enormous interest particularly for the formulation of poorly soluble drugs and vitamins. There are innumerable publications describing its use in different drug formulations and technologies. This review will focus mainly on chemistry and applications in oral and parenteral drug formulations. More specifically, this article will shed light on the utilities of Kolliphor® HS 15 in design and development of self-emulsifying systems, lipid nanoparticles in oral and parenteral applications, and create an understanding of its underlying mechanisms pertaining to its interaction with Pgp with respect to other solubilizers. The regulatory and toxicological information with future perspectives will also be discussed in this review.
It is to be noted that the name Kolliphor® HS 15 is kept in the body of the manuscript while the name Solutol® HS 15 has been left unchanged where indicated in the references.
Chemistry and Physicochemical Properties
As shown in Figure 1, Kolliphor® HS 15 is synthesized by reacting 12-hydroxystearic acid (I) with ethylene oxide (II) in presence of an alkaline catalyst to yield the major components, polyethoxylated derivatives III and V, which represent mono- and diesters. The minor components VII (free polyethylene glycol), IV and VI are also formed.
Quantification of Kolliphor® HS 15
Physicochemical attributes of Kolliphor® HS 15 follow both the USP and Pharm. Eur. compendia. The test methods have been developed and validated per the monographs’ requirements. Though these methods have been used for qualifying the excipient itself the challenges remain to quantify Kolliphor® HS 15 in drug formulations. This is due, in part, to the structural complexity of ethoxylated fatty acid derivatives and because of challenges to separate and identify each of the individual components in the product itself and even more complex in drug formulations due to presence the of many other excipients together.


The distribution of these hydrophilic and lipophilic components in Kolliphor® HS 15 can lead to a greater impact on the stability and the performance of drug products in general. To address this, Janet et al. and Hammond et al. developed an advanced analytical technique using matrix-assisted laser desorption/ionization (MALDI) technique coupled with an ion mobility device to separate and quantify the individual components of Kolliphor® HS 15 and used it as a tool to control the product’s quality attributes.8,9 Zhang and co-workers, developed an analytical method for simultaneous quantifi cation of Kolliphor® HS 15 (lipophilic fatty acid esters, and hydrophilic PEG fragments) in a drug formulation prepared with another excipient like Miglyol® 812.10 With the exception of quantifi cation of only free PEG (ca. ~30%) by HPLC, the lack of compendial test methods, makes it challenging to quantify the main mono- and di-ester moieties in the formulation. The authors developed an UPLC method coupled with a NQAD nebulizer probe for simultaneously quantifying the lipophilic moieties as well as the hydrophilic free PEGs in the formulation.10 The UHPLC coupled NQAD method is unique and capable to separate and quantify both the lipophilic and hydrophilic components without derivatization. In brief, utilizing the UPLC/NQAD technique, the authors quantifi ed the lipophilic component amounts to 67.8% eluted at retention time of 2 min; while the other three hydrophilic PEG components amount to 14.7%, 10.7% and 6.9% eluted at the retention time of approx. 6 min, 7 min and 8.6 min, respectively. The total free PEG component was estimated to be 32.3%, which was in agreement with the compendial acceptance criteria of 27.0% to 39.0% for free PEG (USP). The lipophilic and hydrophilic components were also analyzed by gel permeation chromatography (GPC), a method that used an isocratic condition to estimate the major and minor components by Coon et.al.11 The authors quantified the lipophilic component to be 70% at retention time of 7 min and the hydrophilic component to be about 30% free PEGs at retention time of 12 min, again supporting that the composition of Kolliphor® HS 15 is highly complex but the results are aligned with the manufacturer’s specifications. The GPC method, however, lacked the quantitative determination of the minor pegylated fatty acid components. It can be taken to suggest that the quantitative analysis of Kolliphor® HS 15 might be challenging, especially, if only smaller amounts of Kolliphor® HS 15 are present in the plasma following oral administration where the bioavailability remains very low (ca. 14%). Bhaskar and coworkers used LC-MS/MS method with an ion suppression effect to quantify the Kolliphor® HS 15 in the plasma samples from rats collected following oral and parenteral administration.12,13 Using this method, the authors identified a total of twelve oligomers with the most abundant ions corresponding to m/z 481, 525, 569, 613, 657, 701, 745, 789, 833, 877, 921, 965 by electrospray ionization, and estimated the concentration by combining all the peak areas for each oligomer. The liquid chromatography/tandem mass spectrometry method thus quantified the lipophilic and hydrophilic components in Kolliphor® HS 15, which was found to be 63.3% and 36.7%, respectively. The authors were also able to establish the pharmacokinetic parameters of Kolliphor® HS 15 as well as of 12 PEG oligomers following oral and intravenous administrations in Sprague Dawley rats by using this state of the art technique.
Solubilization Capabilities
Kolliphor® HS 15 is soluble in aqueous media and polar organic solvents but insoluble in apolar solvents. In aqueous solutions, however, it spontaneously forms micelles which helps encapsulation and increases solubility, an attribute dependent on the solubilizer concentration. Figure 2 illustrates that the solubility of poorly soluble drugs increases with increasing amounts of Kolliphor® HS 15. For example, with increasing concentrations of Kolliphor® HS 15 from 1%, 5% to 10%, the solubility increases almost linearly and the micellar size changes only slightly as more drug molecules get encapsulated into the interior core of the micelles.1,14,15
The linear increase at lower concentrations is typical for this kind of solubilizer as polysorbate 80, Kolliphor® EL or Kolliphor® RH 40 also show a similar behavior.15 A further increase of the solubilizer concentration can change the micelle structure and subsequently results in a deviation from the linear profile.

Figure 3 exhibits the solubilization capacity of Kolliphor® HS 15 for a number of model drugs and compares it with other commonly used solubilizers. The data suggests that Kolliphor® HS 15 is an equally effective solubilizer as Kolliphor® RH 40 and polysorbate 80. Out of 10 drugs screened, it was observed that four drugs were best solubilized by Soluplus®, but Kolliphor® HS 15, Kolliphor® RH 40 and polysorbate 80 were found to outperform the others. The ranking of poloxamer 407 (Kolliphor® 407) was not as good as the other solubilizers tested which can be attributed to the much lower amphiphilicity of poloxamers. The difference in hydrophobicity between a fatty acid and the PEG moiety is much larger than between a polypropylene glycol and a polyethylene glycol moiety.
Application Relevant Characteristics
Kolliphor® HS 15, one of the few excellent solubilizers, has widely been used in recent times to improve the solubility of many poorly soluble compounds.4 In comparison with other known solubilizers including poloxamers, polysorbates, and castor oil based surfactants, it shows an exceptional solubilization capability for many insoluble compounds as shown in Figures 2 and 3.

The physicochemical properties of Kolliphor® HS 15 as a solubilizer have been studied extensively.7 With its profound safety and toxicological profile relatively better than polysorbate 80 and/or Kolliphor® EL, it is an excellent choice for screening and pre-clinical pharmacokinetic studies of cytotoxic and oncology drugs among many other indications. Being versatile and highly functional, it offers unique benefits due to its compatibility in in vitro testing as well as in drug formulations as selfemulsifying/micro-emulsifying systems, lipid nanoparticles for oral, parenteral, topical, biologics, nasal, and gene delivery applications.16 Furthermore, its unique structure with availability of terminal free hydroxyl moieties can be modified to a copolymer for safe application in gene delivery. For example, Yin et al. used a Kolliphor® HS 15 grafted copolymer with polyethyleneimine (PEI with Mw 25 kD) in gene delivery to study the cytotoxicity (IC50) against different cell lines, and found that transfection efficiency of graft copolymer (Solutol-g-PEI) was significantly improved owing to its higher complexing ability with DNA compared to pure PEI polymer.17

The micellar structure of Kolliphor® HS 15 helps solubilize the compounds in the micellar core almost independent of pH or biorelevant media. The critical micelle concentration is 0.06-0.1 mM and hydrodynamic radius is 11-15 nm.18 The micelles are stable upon steam autoclaving at 121°C/20 min, making it compatible to heat sterilization procedures, a hallmark of stability of the molecule under harsh conditions.
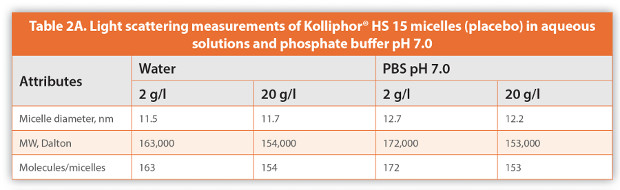
Thysen examined the physicochemical attributes of Kolliphor® HS 15 with and without vitamin E acetate before and after sterilization by autoclave.14 The author did not observe any changes in the particle size of the micelles and/or saponifi cation and pH values of placebo or vitamin formulation. In another study, for example, 1% propofol was eff ectively solubilized with 8% Kolliphor® HS 15, and the formulation was stable and particle size remain unchanged at 40°C for over 8 weeks.19 In an earlier study, it was also observed that solubility of many drugs improved dramatically and drug loaded micelles were also stable for several weeks without any change in the particle size.1 The authors used a CONTIN software program to calculate the size, distribution and composition of the micellar diameter, and measured the molecular weight of the micellar structure. The average MW of a 12 nm Kolliphor® HS 15 micelle comprised of about 170 molecules was determined to be 170 k Dalton. Depending on the type of poorly soluble actives and their concentration the number of active molecules per micelle varied between 22 and 2061, as shown in Table 2. This study provides a deeper insight into the structure of non-loaded and drugloaded micelles.
The amphiphilic characteristics of Kolliphor® HS 15 are also important to form tiny oil droplets from a self-emulsifying drug delivery system (SEDDS/SMEDDS) to carry the maximum payload and effectively deliver the drug molecules.
Special Dosage Forms and Applications
Self-emulsifying drug delivery systems (SEDDS)
Self-emulsifying drug delivery systems (SEDDS) have been a subject of continued interest, due, in part, to faster development of new chemical entities to market as compared to other solid dispersion and micronization technologies.20 Thus, the norm of developing a self-emulsifying formulation requiring solubilizers like non-ionic Kolliphor® HS 15 is highly sought after due to its compatibility with both weakly acidic and alkaline drug molecules (unpublished data). A relatively large percentage of lipophilic (PEG-ylated fatty acids) and substantial amount of hydrophilic or free polyethylene glycol components in Kolliphor® HS 15, play a crucial role in encapsulation and self-emulsification processes with other lipids/oils. For example, Kolliphor® HS 15 in combination with desired amounts of co-surfactants and/or oils enables simultaneously dispersed droplets and maintains supersaturation, that could yield indiscriminate permeation and absorption of drugs through GI tract. The smaller particles created by using the larger amount of the lipid surfactants and cosurfactants in turn help the rapid absorption and enhancement of bioavailability. Patel et al., investigated lumefantrine (LF) SEDDS prepared with the surfactant Kolliphor® EL and oleic acid (OA). The faster dissolution of lumefantrine was attributed to a reduction in droplet size and a greater surface area of the self nano-emulsifying LF-OA ionic complex.21 The faster dissolution was due to significant reduction in diffusion layer thickness caused by nanosizing of particles.22
Selection of the appropriate solubilizers, cosolubilizers and oils is important to design an effective oral drug delivery system with controlled particle size, polydispersity, and zeta potential.23 The authors detailed the identification of lipids and lipid-based excipients for formulations to optimize the oral delivery of lipophilic drugs via SEDDS/SMEDDS. Li and Zhao and Sapra et al. proposed the strategies for early phase screening of SEDDS formulations involving the solubilizers like Kolliphor® HS 15 among others.24,25 The authors also described a range of important routes of administration and the respective excipients/solubilizers because of their abilities to self-emulsify APIs to achieve the desired particle size for required doses, efficacy and stability. Sadurni et al. designed nano-emulsions, each comprised of binary mixtures of Kolliphor® HS 15/Miglyol® 812, Kolliphor® HS 15/soybean oil and Kolliphor® EL/Miglyol® 812, and characterized them by the small angle x-ray scattering and dynamic light scattering techniques.26 The authors noted that the increasing amounts of surfactants between 60% and 90% in SEDDS led to smaller particles (ca. < 50 nm), yielding the distinct phase behavior changes in hexagonal phase (HII) and lamellar crystalline phase (Lα). Heshmati et al. investigated the emulsifying characteristics and pharmacokinetics of four indirubin E083 loaded self-emulsifying systems, each comprised of Kolliphor® HS 15 with varied amounts of medium chain mono- and diglyceride oils, and a cosolvent PEG 400 or ethanol.27 The authors concluded that the higher amounts of the surfactant Kolliphor® HS 15 in SNEDDS, showed a significantly greater bioavailability with respect to SEDDS, which comprised of lower amount of Kolliphor® HS 15, suggesting further that smaller particles are critical to faster absorption and improved bioavailability. Zhou et al. used Kolliphor® HS 15 in design and development of brusatol loaded SEDDS consisting of a medium chain triglyceride (MCT) and polyethylene glycol 400 (PEG 400) as a solvent, and evaluated against dextran sodium sulfateinduced ulcerative colitis.28 The authors observed that the therapeutic efficacy of drug loaded SEDDS improved and bioavailability increased >2-fold as compared to suspension formulation in mice due to the particle size differences.
Particle size reduction is important in aiding the absorption and bioavailability but it also improves the safety of APIs as well as minimizes the adverse effects by avoiding excessive dosing. Benbrook et al. used Kolliphor® HS 15 in the chemoprotective activities of SHetA2 in self-emulsifying systems because of its excellent safety profile and the lack of toxicity when administered by oral gavage to rats.29 The studies concluded that the SHetA2 administered orally at 750 ppm in Kolliphor® HS 15 SEDDS for over 6 weeks, achieved the desired absorption of drug levels without causing any toxicity in mice. At higher dose, however, it accumulated in the lower GI tract resulting in the reduced systemic absorption or poor bioavailability. Menon et al. also used Kolliphor® HS 15 in nanoprobes for cellular and molecular imaging analysis by employing Nile red doped hexamethyldisiloxane (HMDSO) nano-emulsions prepared by Kolliphor® HS 15-lecithin binary combination. The nano-probes had the average particle size of 71±39 nm and were stable in human plasma much longer than 24 hours that enabled to successfully measure the pO2 changes in cells by magnetic resonance imaging.30
Solid Self-emulsifying Drug Delivery Systems (s-SEDDS)
As the interest in self-emulsifying systems continues to grow, so is the interest in solid-SEDDS to counter the shelf life and mitigate the food effects and/or concerns of stability with liquid SEDDS.31 Thus, the efforts continue to develop stable solid-SEDDS for oral dosages to effectively deliver the drugs in pellets and tablets to overcome the precipitation and enhance the stability of drugs in the formulations. Abdalla and Mader used Kolliphor® HS 15 with glyceryl monostearate (GMS) and microcrystalline cellulose (MCC) for stable SEDDS pellets and studied the release profile of the model drug diazepam from the pellets.32 The authors concluded that the diazepam loaded MCC pellets with Kolliphor® HS 15, formed a self-emulsified system upon contact with aqueous media, and drug release was expedited dramatically as compared to those lacking Kolliphor® HS 15. These pellets showed a sluggish release and did not perform well. This study also sheds light on how the solubilizers like Kolliphor® HS 15 embedded in the solid MCC matrix had the ability to quickly self-emulsify and strongly improve the delivery of poorly soluble molecules. Other groups also used solid matrices like Neusilin US2 and mesoporous silica, and developed the s-SEDDS with aims to increase solubility and enhance bioavailability of drugs.33,34 Sri et al. evaluated the s-SEDDS formulation of valstaran comprised of Kolliphor® HS 15, PEG 400 and Capmul® MCM adsorbed on Neusilin® US2, and investigated the dissolution and pharmacokinetics of the drug from a solid SEDDS adsorbed matrix. The bioavailability of valsartan in rats was improved 1.6-fold as compared to a suspension.35 Cerpnjak et al., on the other hand, used adsorbing polymeric matrices each composed of HPMC or maltodextrin with Kolliphor® HS 15 as solubilizer with another co-surfactant/solvent in design of self-emulsifying systems for naproxen prepared by a spray drying process.36 The authors observed that maltodextrin based s-SMEDDSs were granular smooth-surface microspheres, while the HPMC based s-SMEDDS were irregularly shaped microparticles. The dissolution profiles of maltodextrin based particles showed a faster release, very similar to liquid SMEDDS, but the HPMC based s-SMEDDS showed an incomplete and slightly longer dissolution profile due to gelling characteristics of this polymeric carrier. Other solid matrices have also been evaluated in lyophilized matrices. For example, Li et al. evaluated the sucrose based lyophilized solid SEDDS formulation of Lornoxicam in rats.37 The solid formulation was prepared by adsorbing a liquid SEDDS consisting of Kolliphor® HS 15, Labrafil® M 1944 CS and Transcutol® HP. The pharmacokinetic parameters were assessed and the bioavailability of the drug improved over 1.5-fold with respect to the commercially available tablets, suggesting that Kolliphor® HS 15 is a an effective solubilizer and suitable for lyophilization with sugars in the development of SEDDS tablets. Kolliphor® HS 15 is compatible with sugars and oleochemicals, it is also compatible with cyclodextrins. For instance, the liquid efavirenz dispersions prepared with Kolliphor® HS 15 and β-CD (2:0.05) increased the absorption rate ~5-fold and the AUC by ~ 2-fold as compared to pure drug. In vitro dissolution of efavirenz increased about 4-fold, while the bioavailability enhanced 17-fold when Kolliphor® HS 15 and β-CD were used with povidone K30 in a ternary mixture as opposed to a binary mixture of β-CD and PVP K30.38
Varshosaz and Ghassami studied fenofibrate in spray dried binary mixtures, each prepared separately with Eudragit® E100 and Kolliphor® HS 15 and HPC as the carriers.39 Colloidal silica was used as an antitacking agent. Fenofibrate was dissolved in acetone with Eudragit® E100 or Kolliphor® HS 15 or hydroxypropyl cellulose (HPC) at 1:1 and 1:5 ratios (drug/excipient) and spray dried with silica as an anti-tacking agent. The dissolution efficiency varied for each binary formulation and was dependent upon the ratios of API and polymeric carrier. The angle of repose or flowability was slightly lower with a binary mixture of Kolliphor® HS 15 and fenofibrate as compared to Eudragit®/fenofibrate binary blend. This difference might be related to the waxy nature of the Kolliphor® HS 15 and its stickiness that decreased flowability and dissolution efficiency as compared to Eudragit® and HPC polymers. The dissolution enhancement rate of the drug with increased flowability was presumably due to particle size reduction of binary powder blends as compared to untreated drug.
Gonzales and co-workers investigated the self-emulsifying properties and pharmacokinetic profiles of cyclosporine A (CyA) in a formulation comprised of Kolliphor® HS 15, Labrafil® M2125CS and oleic acid (/7:2:1 (v/v/v)).40 The oral bioavailability of these SEDDS in rats was about twofold greater than the micronized suspension lacking Kolliphor® HS 15 (70% vs. 36%). The improved oral bioavailability was caused by marked improvement in the solubility of CyA in microemulsion as compared to micronized suspension (ca. 136 μg/ml vs. 23.2 μg/ml in human gastric juice, and 133 μg/ml vs. 10.8 μg/ml in simulated intestinal juice). It also holds true for many other drugs in SEDDS/SMEDDS. For example, Cui et al et al. investigated in vivo efficacy of a drug in rat of a formulation containing 50% Kolliphor® HS 15 as a surfactant and 35% Transcutol® as a co-surfactant and 15% of an oil consisting of ethyl oleate, Maisine® 35-1, and Gelucire® 44/14. The bioavailability of the drug in SEDDS improved > twofold over the tablet formulation.41
Lipid nanocapsules, solid lipid nanoparticles and nanostructure lipid carriers
Kolliphor® HS 15 has been investigated for in vitro hemolytic activity of a micro-emulsion formulation containing oleic acid and soybean oil.42 It was found that the Kolliphor® HS 15 based micellar nanoparticles were stable over 3 months and were far less hemolytic (<0.2%), making it most suitable to parenteral formulations without any additional cosolvents. Chauvet et al. found that Kolliphor® HS 15 based lipid nanocapsules (LNCs) bearing the particle size of 25 nm, influenced cellular calcium signaling in the brain through activation of neuronal channels, presumably by interaction of cholesterol rich membrane lipids with lipophilic/fatty acid chains of the solubilizer.43 Such interactions could have important clinical implications as these LNCs are able to penetrate deeper into the membrane, allowing to endocytose the drug into the cells, and affecting the biological activities.44 Dulieu and Bazile developed the lipid nanocapsules (LNCs) from freeze dried formulations containing Kolliphor® HS 15 and Lipoid® S100 in trehalose solution.45 The resulting particles were stable due to a lipid protective coating around the emulsion droplets, and surrounding Kolliphor® HS 15 on the outer core of the particles, providing a stealth protection. The rigidity and stability of the core increased as the ratios of lipid/Kolliphor® HS 15 increased at the outer coating surface. This stealth protection is important for longer systemic circulation of a drug as it gets carried through for delivery into the specific tissues or site.46
Pitorre and co-workers studied the lipid nanocapsules (LNC) derived from Kolliphor® HS 15, Labrafac® W and Span® 80 to target the lymph nodes by subcutaneous administration of a model compound.47 The authors demonstrated that when the fluorescent labeled compound was administered in rats subcutaneously (sc) in neck versus the IV in tail vein, it was highly selective to only specific lymph nodes but was less selective and poorly distributed in all other lymph nodes. The particle size of LNC (ca. 50 - 100 nm) was apparently important in distinguishing the bio-distribution and absorption of these nanocarriers by the tissues or organs.48 Drug loaded LNCs, and their escape by RES and being recognized for uptake by the cancer cells to help deliver the drugs at the specific target sites by subcutaneous injection as opposed to intravenous route, provide an alternative and effective delivery of drug while reducing the adverse effects. Paclitaxelloaded LNCs comprised of Kolliphor® HS 15, cholesterol, Labrafac®, and Lipoid® S100 were used in vitro and in vivo efficacy studies of drugs against multidrug resistance (MDR) glioma tumors and found that the Kolliphor® HS 15 containing LNC loaded with paclitaxel demonstrated superb activities against commercially available Taxol® formulation containing Kolliphor® EL and ethanol.44 These differences in bio-efficacy were due to tumor targeting through intracellular compartmentalization of paclitaxel in the multi-drug resistance (MDR) cells caused by interaction and inhibition of membrane associated glycoprotein (Pgp) efflux pump, leading to cell death. LNC loaded paclitaxel suspension comprised of Kolliphor® HS 15, Lipoid® S100, and Captex® 8000 have also been evaluated for increasing the oral bioavailability of paclitaxel.49 In this study, the authors observed that LNCs were highly effective in enhancing the bioavailability to 21%, about a 3-fold increase as compared to Kolliphor® EL based Taxol® formulation (approx. 7%). Like LNCs, the self-emulsifying loaded paclitaxel formulations, s-SEDDS and s-SMEDDS, were equally effective and showed, respectively, 5-fold and 2-fold increases in the oral bioavailability as compared to Kolliphor® EL based Taxol® formulation.50,51
Improving the bioavailability of paclitaxel loaded LNC with Kolliphor® HS 15, offers a unique opportunity to further explore the structural components and packing of lipids in these assemblies. To shed light on the understanding of physio-chemical and morphological characteristics of lipid nanoparticles derived from glyceryl tristearate/castor oil and prepared by combining each with a solubilizer like Kolliphor® HS 15 or poloxamer 188, Dora et al used the dynamic light scattering (DLS), differential scanning calorimetry (DSC), wideangle X-ray scattering (WAXS), atomic force microscopy (AFM) and transmission electron microscopy (TEM and cryo-TEM have been used to elucidate the structures of these assemblies.52 The oil/water (o/w) nano-emulsion (NE) and solid lipid nanoparticle (SLN) were derived from the solubilizer with castor oil or tristearin. Lipid-based nanocarriers (NLCs) were prepared by hot solvent diffusion method by dissolving castor oil, tristearin, and lecithin in organic solvents followed by homogenization of solution containing 0.1% Kolliphor® HS 15 (Tm 24.3 °C; ΔH 78.3 g/J) or 1% poloxamer 188 (Tm 54.3 oC, ΔH 129 g/J). In a separate study, the authors observed that quercetin loaded SLNs each with poloxamer 188 and Kolliphor® HS 15 solubilizers exhibited 45% and 70% release, respectively; while the NLC did not control the release because of the castor oil was not internalized and surrounded by the solid lipid.53 Joshi et al. investigated the delivery of a poorly soluble anti-malarial drug artemether (ARM) in the nanostructured lipid carriers (NLC) as the Nanoject for intravenous delivery in rats, and observed an extended release of drug over 20 hours as compared to 6 hours with the conventional micro-emulsion formulation.54 The Nanoject’s extended characteristic was due to presence of the saturated glyceryl dilaurate lipid that controlled the API’s release and improved the adverse effect and safety. As a consequence, the survival rate with NLC IV administration improved as compared to marketed oily intramuscular formulation (Larither®). The data also suggests that the Nanoject, an extended NLC formulation, significantly improved the ARM’s antimalarial activity lasting over 20 days as compared to intramuscular (IM) dose formulation. Furthermore, the biocompatibility and microstructure formation with excipients like Kolliphor® HS 15 in the Nanoject rendered the reduction in pain upon intravenous injection. Because of their excellent safety in oral and injectable formulations, and mildness to skin, the SLN and NLC have also been investigated in the topical drug delivery.55,56
Liquid vs. Solid dispersion formulations
Lipid based self-emulsifying and solid dispersion technologies are competing technologies for increasing solubility and enhancing bioavailability of the same poorly soluble molecules. The choice of the excipients though is different for each technology, and may vary depending upon the formulation and dosage requirements, and the API per se. El-Laithy and co-workers evaluated two poorly soluble molecules biphenyl dimethyl dicarboxylate and silymarin in two different formulations, one based on solid dispersions (SD) prepared with poloxamer 188 by melting and the other by coprecipitation with deoxycholate, and compared the two amorphous solid dispersions (ASDs) with respect to a lipid based self-emulsifying SMEDDS comprised of 40% polysorbate 80, 10% Miglyol® 812 and 50% Transcutol®.57 The authors observed that both formulations, SMEDDS as well as ASD, were equally effective as the commercially available product against the hepatic fibrosis in Albino rats. Examples like these in the commercial world are limited; products like lipid based ritonavir (Norvir®) and ritonavir and lopinavir (Kaletra®), launched originally as soft gel capsules (SEDDS) and substituted later on by amorphous solid dispersions (ASD) tablets because of much higher stability and active contents. Each dosage though requires different excipient compositions in the individual formulations and technologies, both SMEDDS and ASD dosages possess similar pharmacokinetics profiles.58 For example, ritonavir’s liquid SEDDS contains polyoxyl 40 hydrogenated castor oil (Kolliphor® RH 40) and ritonavir/lopinavir’s liquid SEDDS contains polyoxyl 35 castor oil (Kolliphor® EL), both having a similar HLB value as Kolliphor® HS 15. Pharmacokinetic profiles and bio-distribution of model compounds in liquid formulations containing Kolliphor® HS 15 and other solubilizers have been investigated following single IV dosing.59 Scheller et al. investigated the delivery of galanin-receptor 3 selective antagonist, SNAP 37889, and achieved the desired solubility in 30% Kolliphor® HS 15 micro-emulsion injectable solution by solubilizing the drug in a paste form and diluting it in phosphate buffer.60 The authors used this IV formulation for evaluation of the galanin antagonist in neuropharmacology of rats and mice. Other applications of Kolliphor® HS 15 include the development of cyclosporine ophthalmic sterile liquid solution with castor oil and glycerol to yield formulations with the excellent stability and efficacy with no apparent toxicity, incompatibility and/or ocular irritation.61 Zhao et al. used a phospholipid based IV micro-emulsions composed of Kolliphor® HS 15, Miglyol® 812, soy lipid, PEG 400 and ethanol in the parenteral delivery of ibuprofen eugenol ester (IEE).62 Solubility of the ibuprofen derivative improved over 21000-fold (64 mg/ml) as compared to placebo, and the ibuprofen based IEE micro-emulsions exhibited prolonged acting time, attributed due to the stealth protection of droplet surface by Kolliphor® HS 15 from being recognized by opsonins, and taken up by the cells of reticuloendothelial systems (RES).63,64
Parenteral Propofol IV formulation
Propofol (2,6-diisopropylphenol) has been formulated in Kolliphor® EL aqueous solution but it brings risk associated with anaphylactoid reactions and high incidence of pain associated with injection.65 To alleviate these side effects, an alternative o/w based 1% propofol emulsion, preferentially formulated with soy oil, egg phospholipids and glycerol, has been developed but due to unforeseeable challenges including the elevated serum triglycerides level and impediment in fat metabolism due to high lipid content in the formulation, it warrants additional studies to develop a much safer IV micro-emulsion formulation. To alleviate much of the safety concerns and mitigate the adverse effects, Kolliphor® HS 15 is an alternative surfactant for the development of micro-emulsion IV formulations.66 The authors used Kolliphor® HS 15 with PEG 400, propylene glycol, glycofurol in a 1% propofol IV emulsion formulation. Ryoo et al. also formulated a 1% propofol IV micro-emulsion with 8% Kolliphor® HS 15 in ethanol and found that it qualified the desired criteria; was stable and safe without being hemolytic to red blood cells. These findings led to the development and marketing of Aquafol®.67 Lee et al. further investigated the safety of propofol IV micro-emulsion formulation and modified the composition containing only 0.7% of Kolliphor® HS 15 mixed with 10% purified poloxamer 188 in 1% glycerin as a cosolvent.68 The re-formulation dramatically improved the safety with fewer adverse effects as compared to the previously marketed Aquafol® formulation containing 8% Kolliphor® HS 15 with 5% glycofural as a cosolvent.67 The adverse effect like pain following injection was evaluated by quantifying the free propofol in propofol IV emulsions. Date and Nagarsenker evaluated the propofol IV emulsion and found that a binary mixture of Kolliphor® HS 15 and polysorbate 80 combined in 1:1 ratio, was more effective as compared to those consisting of cosolvents like glycofurol and/or polypropylene glycol.69 Furthermore, the Kolliphor® HS 15/polysorbate 80 binary combination of propofol IV emulsion drastically improved safety and potentially reduced pain on injection. It also provided a better content uniformity without compromising the pharmacodynamic activity and physicochemical stability with respect to commercially available Propovan® emulsions. It is also worth noting that the free propofol, if left non-encapsulated in micro-emulsion caused bearable pain following IV injection but its severity was wide spread in twice larger population as compared to medium-chain or large-chain triglyceride based propofol treated IV micro-emulsion formulation.70
A few other propofol IV micro-emulsions are in clinical stages of development. In a comparative clinical study, propofol IV Kolliphor® HS 15 formulation was evaluated with Kolliphor® HS 15 in combination with soy lipid based nano-emulsion and found that both formulations performed equally good with superb safety and efficacy with fewer side effects in patients.71 Rittes et al. also investigated the Kolliphor® HS 15 in propofol IV nano-emulsion and compared it with soy lecithin IV emulsion formulation in patients undergoing gynecological procedures. The patients were monitored for heart rate, systolic and diastolic blood pressures and peripheral oxygen saturation.72 The authors concluded that the groups with 1% or 2% propofol in soy lipid based and Kolliphor® HS 15 surfactant based IV nano-emulsion formulations elicited pain on injection but the intensity of pain was less severe with Kolliphor® HS 15 IV, and both formulations showed neither hemodynamic changes nor any adverse eff ects of clinical relevance. It was also consistent with the findings that the adverse reaction was not related to Kolliphor® HS 15 but due to free propofol if left un-encapsulated.73 In a clinical study, the non-lipidic Cleofol® IV formulation showed much greater incidence of severe pain as compared with medium chain triglyceride based Lipuro® IV formulation and long chain triglycerides based 1% propofol IV emulsion available as Diprivan®. This adverse eff ect was attributed to free propofol in Cleofol® IV formulation which lacked the medium or long chain triglycerides.74 To alleviate the adverse eff ect associated apparently with free propofol in IV micro-emulsions, Morey et al. used sodium salt of fatty acids (C8, C10, C12) in combination with purified poloxamer 188 for complete encapsulation of drug to yield a transparent micro-emulsion solution with particle size < 50 nm.75
Inhibition of Pgp efflux pump
P-glycoprotein (Pgp) (MW 170 kD), is a large integral transport protein in the membrane when overexpressed pumps out the undesired molecules entering and/or accumulating into the cells. Thus, the overexpression of Pgp provides a protective function in blood brain barrier, colon, pancreas, kidneys, and gastrointestinal tract, more specifically, it prevents the accumulation of xenobiotics and metabolites in the brain. In addition, the overexpression of Pgp is highly prevalent in the cancer patients undergoing chemotherapy.76 There is a range of chemicals and therapeutic agents which act as chemosensitizers or substrates that affect the function of Pgp pump.77 For instance, surface active agents such as poloxamers have shown different activities on the Pgp efflux depending upon the structure of the molecules.78 Batrakova et al. classified the poloxamer into 4 groups and those with longer hydrophobic polypropylene oxide (PPO) moieties penetrate deeper into the blood brain barrier (BBB) membrane leading to endocytosis and accumulation of drug into the cytoplasm.79 Constantinides and Wasan also investigated the Pgp inhibition by lipid based excipients such as Kolliphor® EL, Kolliphor® HS 15, poloxamers among others with poorly soluble drugs and found a significantly improvement in intestinal absorption of these molecules due to Pgp inhibition, which probably led to acceleration of drug transport across the cell membrane.80

Wang and co-workers studied the mechanism of Pgp activities on mouse fi broblast cell line NIH 3T3 of a number of surfactant/lipid based solubilizers acceptable to intravenous (iv), intramuscular (im) and oral (p.o.) formulations.81 The Pgp inhibitory activities data normalized against 0.9% NaCl solution, is summarized in Table 2.
The data clearly indicates that the solubilizers tested had a very different impact on Pgp inhibition ranging from no eff ect to a strong one. For instance, Kolliphor® HS 15 resulted already at a very low concentration of 8 μg/ml in a normalized IC50 of 4.2 which was only exceeded by TPGS with a value of 6.8. In contrast most of the poloxamers except poloxamer 124 did not exert a strong action, even not at higher concentrations. In general, the lipid based surfactants were more potent regarding Pgp inhibition than poloxamers. With reference to cyclosporine A, the activities of the best solubilizers were still below the drug’s Pgp inhibitory property.81
The mechanism by which the lipid based solubilizers like Kolliphor® HS 15 act as chemosensitizers that infl uences the Pgp inhibition is poorly understood. Such inhibitory activity is apparently critical for explaining the permeation of drugs via endocytosis and, hence, the efficacy thereafter. Kolliphor® HS 15 based CriticalSorb® was used as an absorption enhancer in the epithelial cell cultures (e.g. A549, Caco-2 and Calu-3), with an aim to deliver or transport the biologic drugs via mucous membrane.18 The authors concluded that Kolliphor® HS 15 promotes transcellular apical to basolateral transport of biologics through Calu-3. Furthermore, Kolliphor® HS 15 when tested against the epithelial cell cultures showed a relatively high IC50 (ca. 6.5–10.2 mM) as compared with other absorption enhancers or surfactants such as sodium cholate (2.04–4.94 mM) and polysorbate 80 (5.41–6.49 mM), suggesting much milder in vitro activities of Kolliphor® HS 15 than other solubilizers. Huo et al. evaluated the mixed micelles of Kolliphor® HS 15 and poloxamer 407 (4:1) for delivery of the oncology drug icariside II (IS) in rats, and found that the mixed micelles improved the entrapped efficiency to 95% and increased >2-fold bioavailability, presumably caused by increasing permeability across cell membrane due to inhibition of Pgp.82 In a study, Kommuru and co-workers demonstrated that Kolliphor® HS 15, when used as a surfactant carrier, may increase the permeability and intracellular concentration and residence time of CoQ10 via paracellular transport by inhibition of Pgp and/or CYP450 enzymes.83 These findings have been further supported by enhanced bioavailability of other drugs that influence the inhibition of Pgp and cytochrome P450 (CYP) 3A.84 Though the specific molecular changes to Pgp are not yet fully understood, the influence of Kolliphor® HS 15 on the Pgp could be non-specific conformational changes due to surfactant molecules penetrating deep into the plasma membrane. Like Kolliphor® HS 15, other lipid solubilizers such as vitamin E-TPGS, Kolliphor® EL, Kolliphor RH 40, and polysorbate 80, are well-known chemosensitizer that may temporarily inhibit the Pgp pump, which could then trigger the transport of drugs across the intestinal mucosal membrane, leading to faster absorption and improved oral bioavailability preferably by lymphatic uptake.85
Biological activities of Kolliphor® HS 15
Shubber and co-workers investigated the mechanism of in vitro mucosal permeability of Kolliphor® HS 15 based CriticalSorb® as absorption enhancer in the epithelial cell cultures of A549, Caco-2 and Calu-3 cell lines, with aims to deliver the biologic drugs via mucous membrane by epithelial cellular transport, a novel route to overcome the parenteral route for administration of macromolecules such as insulin, hGH, albumin and IgG.18 The authors demonstrate that increasing the MW of macromolecules slowed down the permeability of these molecules, while the permeation of Kolliphor® HS 15 was apparently faster with low MW macromolecules. The permeation of drug molecules through Calu-3 cell lines decreased in the order: Insulin > hGH > Albumin > IgG. In another unrelated study, Lin et al. demonstrated that the fatty acid methyl esters of palmitic (C16:0) and stearic (C18:0) acids exhibit the potent vasodilatory properties which could be important in neuroprotection following cerebral ischemia.86 The authors compared the biological activities of Kolliphor® HS15 with the structurally similar fatty acid methyl esters having an ester linkage with C16 and/or C18 fatty acids. The authors demonstrated the neuroprotective effect by calculating the number of survived hippocampal neurons by individual expression of enhanced levels of Kolliphor® HS 15, palmitic acid methylester, and stearic methyl ester in treated rats as compared to controls. The data suggests that with appropriate dosage of Kolliphor® HS 15 and/or C16 and C18 methyl esters, the levels of each component was overexpressed in neurons with all showing the increase of about 68-69%, which was an effective level against cerebral ischemia treatment. In a study Bartels et al., investigated the protective effect of antioxidative vitamins against lipid peroxidation by parenteral administration of Kolliphor® HS 15 formulations comprised of α-tocopherol, ascorbic acid, and β-carotene on the extent of ischemia and reperfusion liver injury.87 In another study, Bartels and co-workers used the soybean lipid in combination with Kolliphor® HS 15 in parenteral infusion of lipid soluble anti-oxidative vitamins to reduce the adverse effects by monitoring the toxicity by the elevated liver enzymes caused by the residual solvents.88 Coon et al. investigated the multidrug resistance of Kolliphor® HS 15 in mice, and found that like verapamil, Kolliphor® HS 15 reduced the efflux of rhodamine through KB 8-5-11 cells but it did not affect the transport of alanine or glucose into the cells, and claimed that its effect on membrane transport activity was highly non-selective. The IC50 of Kolliphor® HS 15, however, was 140 μm as versus 65 μm of verapamil against the KB cell line.11 Buckingham et al. investigated the PEG-fatty acids esters in vitro performance against KB cell lines. The optimized oleic acid ester with 20 mole ethylene oxide (or PEG), e.g. CRL 1337, was over 8‐fold as potent as Kolliphor® HS 15 and over 19‐fold as potent as Kolliphor® EL in vitro, as evident by accumulation of rhodamine 123 pigment in multidrug‐resistant KB 8–5–11 cells.89 The authors concluded that the hydrophobic fatty acid components of surfactant and its hydrophilic‐lipophilic balance are critical in determining the potency of a surfactant. Woodburn and co-workers investigated the binding of lipoproteins (LDL and HDL) and porphyrin based photosensitizers (C8KC and MP) with Kolliphor® HS 15 and compared the binding assay with a synthetic solubilizer/emulsifier CRM, comprised of poly-(oxyethyleneglycerol) triricinoleate bearing the structurally similar hydrophobic and hydrophilic components as the Kolliphor® HS 15.90 The authors observed that Kolliphor® HS 15 and CRM when used as vehicles showed similar interactions with C8KC and MP in the in vitro electrophoretic assay. Furthermore, higher concentrations of Kolliphor® HS 15 and CRM (>0.06%) led to a decrease in electrophoretic mobility of human LDL and HDL in vitro. In mouse, both Kolliphor® HS 15 and C8KC showed a similar half-life (t1/2) of 1.7 hours, and were cleared out from mouse plasma in vivo over 6 hours. It can further be taken to suggest that vehicles like Kolliphor® HS 15 were able to circulate for much longer period alongside the drug in the plasma without being taken up by RES.64
Compatibility with Plastic Containers
Kolliphor® HS 15 is an exceptional solubilizer for use in parenteral formulations due to its controlled manufacturing particularly regarding microbial and endotoxin contamination. The storage of Kolliphor® HS 15 in plastic containers or bags may lead to degradation or erosion of the storage reservoir caused by extraction of low molecular weight components. Thus, the attempts have been to minimize the leaching and extraction of additives from the package liner.91 Only limited information is available to demonstrate the suitability of the aqueous micro-emulsion Kolliphor® HS 15 formulation in such containers. Cheung et al. investigated the leachable and extractable properties of commonly used solubilizers from iv-bags and iv-infusion lines derived from polyvinyl chloride (PVC) plasticized with 30-40% of bis-(2-ethylhexyl) phthalate (DEHP) as an additive. The PVC/DEHP IV bags each contained Kolliphor® HS 15 (5 mg/ml), polysorbate 80 (3 mg/ml), polysorbate 20 (3 mg/ml), or poloxamer 188 (3 mg/ml) at pH 7.92 The authors observed that the leaching effect was insignificant or none with poloxamer 188 but all other solubilizers including PS 80, PS 20 and Kolliphor® HS 15 resulted in leaching of DEHP. The leaching ability increased linearly as the solutions in the IV bags were exposed to longer periods at similar concentrations and usage conditions. A similar trend was also observed with PVC IV bag/infusion tube plasticized with tri-2-ethylhexyl trimellitate (TOTM) as an additive. Like PVC/DEHP IV bags, with PVC/TOTM IV bags the leaching ability decreased linearly in the order: Kolliphor® HS 15 >> polysorbate 80 > polysorbate 20 >> poloxamer 188, suggesting that poloxamer 188 outperformed in comparison with other solubilizers such as polysorbate 80, polysorbate 20 and Kolliphor® HS 15. This is not surprising since poloxamer 188 has a much lower solubilization capacity which counts also for extraction of lipophilic plasticizers. This study also sheds light that Kolliphor® HS 15 may not be compatible with the PVC IV bags containing DEHP and TOTM as additives. Since polysorbate 80 and polysorbate 20 are commonly used to stabilize the proteins and large molecules, the leaching abilities of the polysorbates could be a disadvantage for anticipated use in the biologics’ IV formulations, however, the typical concentrations used here are much lower. In this regard poloxamer 188 in general due to its compatibility with the plasticizers in PVC IV bags, should be more suitable which may accelerate the usage of poloxamer 188 as a stabilizer downstream in injectable IV and infusion formulations, and as a shear protector in the upstream cell culture.93

Regulatory and Toxicological Perspectives of Kolliphor® HS 15
Kolliphor® HS 15 is monographed in the United States Pharmacopeia by Polyoxyl 15 hydroxystearate, and the European pharmacopeia by Macrogol 660 hydroxystearate. It is also listed in the inactive ingredient database (IID).94
Safety and toxicology of Kolliphor® HS 15 have been evaluated in nonclinical studies and the results have been reported.95 These studies aid in the development of formulations with the guidance on usage levels for certain routes of administration. Several drug products containing Kolliphor® HS 15 have been approved globally to date.
Briefly, Kolliphor® HS 15 does not show significant signs of acute toxicity in rats, mice, dogs and rabbits as shown by studies with oral and parenteral administration. The oral LD50 is >20 g/kg in rats and mice. The acute intraperitoneal LD50 is >8 g/kg for mice, and the intravenous LD50 for rats and rabbits is >1 g/kg. In beagle dogs, the intravenous LD50 is >3 g/kg, however, symptoms of histamine release were observed in a dose dependent manner, but this sign was reversible within a few hours without causing any mortality or death. Histamine release in dogs as the most sensitive animal is a general side-effect of solubilizers with a high amphiphilicity and solubilization capacity. Thus, it occurs also with polysorbate 80 and Kolliphor® ELP, typically to a much larger extent.95
In sub-chronic tox studies, the oral dosing of Kolliphor® HS 15 in dogs continued for 4 and 13 weeks respectively at 1g kg body weight, did not cause any specific toxicity and histopathological changes or organ toxicity or any signs of histamine release (NOAEL >1g/kg). Similar studies were performed in rats resulting in NOAELs of 2g/kg and >1.5g/kg. Repeated intravenous administration continued for 13 weeks at 750 mg/kg in rats, also did not show any specific sign of toxicity; the caused lipid deposition in liver and spleen tissues was considered not to be an adverse effect. Other studies lasting 2 and 4 weeks revealed a NOAEL of >200 mg/kg, the highest dose tested. Dogs are much more sensitive to parenterally applied compounds and thus a dose of 100 mg/kg caused pseudo-allergic reactions associated with histamine release, however no other signs of substance specific toxicity or histopathological changes were observed. No pseudo-allergic reactions occurred at a dose of 25 mg/kg. In comparison, Kolliphor® EL at 5 mg/kg caused the allergic reaction and the effect was equivalent to 100 mg/kg dosing of Kolliphor® HS 15, suggesting that the latter has a tremendous benefit in this regard. Kolliphor® HS 15 was compared in another i.v study in dogs at 100 mg/kg with polysorbate 80 resulting in much lower histamine levels, as shown in Table 3.96 Taking these results into account, Kolliphor® HS 15 represents the safest excipient in the field of strong solubilizers.7
Kolliphor® HS 15 did not show any sign of genotoxicity either in Salmonella typhimurium reverse mutation assay (Ames test), chromosome aberration test, HPRT test or mouse micronucleus test. The reproductive and developmental toxicity in rats at a maximum intravenous dose level of 464 mg/kg did not reveal any substancerelated influence on the prenatal development of the embryo or fetus, or postnatal growth.
From drug formulation perspective, Kolliphor® HS 15 has been evaluated for cancer preventative activity of a new chemical entity SR13668 in dogs and monkeys.97 SR13668 formulation did not show any pathological changes including hematology, clinical chemistry and coagulation effects. The bioavailability of drug following dosing p.o. at 90 mg/kg, showed an increase to about 15% and 7% in dogs ad monkeys, respectively. These findings were critical in assessing the clinical studies of SR13668 in humans. Reid et al. investigated the Akt inhibitor SR13668 in Phase 0 clinical chemoprevention trial in healthy volunteers, and observed that formulation containing Kolliphor® HS 15 with TPGS yielded the maximum oral bioavailability as compared to PEG 400/Labrasol® formulation.98 From the pharmacokinetic findings, the authors concluded that the Phase 0 data could be used as a guidance for the initiation of Phase 1 clinical studies. Dulin evaluated the Kolliphor® HS 15 and other solubilizers in early phases of development in oral self emulsifying system (SEDDS) and found that Kolliphor® HS 15 formulation outperformed and improved the solubility and bioavailability of a new chemical entity (NCE); which necessitated the advancement of a lead candidate in the clinical development.99 This study showed that the oral bioavailability improved several folds with Kolliphor® HS 15 as compared with phospholipid formulation due to much slower rate of absorption with longer Cmax in early stages of screening with lipids in animal models. Consequently, the NCE in Kolliphor® HS 15 formulation further advanced to the first-in-human Phase 2 studies after successful Phase 1. In all 12 subjects studied, Kolliphor® HS 15 formulation was safe and caused no adverse effects after first dosing at 150 mg/capsule and subsequently dosing to maximum tolerated dose (MTD) of 1500 mg/10 capsules, demonstrating an excellent tolerability and safety of Kolliphor® HS 15 in the formulation.100 Stokes et al. conducted separately toxicological studies of Kolliphor® HS 15 and Kolliphor® RH 40 formulations each with polyethylene glycol 400 as a co-solvent in rats and dogs at different dose volumes, at 10 mL/kg for rats and at 2 mL/kg or 5 mL/kg for dogs.101 The authors observed that neither vehicle, regardless of dose volume, nor formulation caused any toxicity either in rats following 91 days of dosing or in dogs following 8 days of dosing. Kolliphor® HS 15 formulation produced only a fewer adverse effects in rats notably at much higher dose volumes, causing only slight dehydration and hemoconcentration. In lower dose volumes, however, both Kolliphor® HS 15 and Kolliphor® RH 40 formulations were safe given orally and minimized the severity or incidence of adverse effects.
Conclusion
Kolliphor® HS 15 is an excellent solubilizer with unparalleled attributes leading to a safe administration of oral and injectable drug molecules. The data presented in the review article and reported independently by many other authors from earlier publications, clearly demonstrates that Kolliphor® HS 15 is highly versatile and is an exceptional excipient/solubilizer for screening of NCEs in the early phases and building platform to provide a good start in leading the way to select the right formulation technologies for development of lead candidates. It also demonstrates that the safety of this excipient is well studied which had helped attain the recognition for freely usages in many approved marketed drug products globally for both in humans and animals as well. With the recent launch of parenteral and ophthalmic drugs in the US, Kolliphor® HS 15 is no longer a novel excipient. As a result, the global acceptance of Kolliphor® HS 15 in future drug candidates will continue to rise. The compliance to the compendial monographs in the USP and European pharmacopeia, further ease the acceptance in the solid and liquid oral SEDDSs/SMEDDS as well as in parenteral formulations. Other applications also continue to take heed such as in biologics by virtues of its structural compatibility with a wide range of lipid and non-lipid based surfactants and solubilizers and polymers alike and with macromolecule APIs such as peptides, nucleic acids and biologics. With an increase in the number of NCEs from the discovery, Kolliphor® HS 15 will continue to play an important role as a solubilizer for the molecules either small or large with difficult-to-dissolve attributes and pave the way in the industry for development of drug candidates by non-conventional novel formulation technologies. The NCEs will dictate the technologies requiring Kolliphor® HS 15, either in liquid oral versus parenteral self-emulsifying and LNC or solid oral based s-SEDDS formulations and efficient delivery of low, medium or high dose drugs.
Author Biography
Shaukat Ali has worked in the pharma industry for over 25 years including 15 years at BASF. His areas of expertise include the controlled release, solid dispersions, lipid based emulsifying systems, liposome drug delivery, and film development technology. He received his PhD in chemistry from the City University of New York, and postdoctoral training from the University of Minnesota and Cornell University. He is the editor-in-chief of Journal Analytical and Pharmaceutical Research and serves as a member of the editorial advisory board of several pharmaceutical journals including the American Pharmaceutical Review. He also serves as a panel of expert in the USP expert committees for General Chapters-Physical Analysis, Continuous Manufacturing and Excipient Performance <1059>. He has published over 45 articles in the scientific journals and is the inventor of 14 US patents.
After having worked for 7 years at Knoll AG in Ludwigshafen, Karl Kolter joined BASF AG in 1993, where he has been responsible for R&D activities in pharmaceutical excipients, drug formulations and the application technology of vitamins and carotenoids for pharma and food. Dr. Kolter’s current work is the development of innovative excipients mainly for solid oral dosage forms, which has already resulted in various new products in the Kollicoat® and Kollidon® range (Kollicoat® MAE 30 D, 100P, SR 30 D, IR, IR White, Protect, Kollidon® SR, CL-F, CL-SF, Ludipress® LCE). He obtained his Ph. D. in pharmaceutical chemistry at the University of Mainz, Germany. He has published more than 100 articles and posters, and is the inventor of 90 patents.
References
- F. Ruchatz and H. Schuch, Physicochemical properties of Solutol® HS 15 and its solubilizates. BASF ExAct, 1998, 1, 6–7.
- R. G. Strickley, Solubilizing excipients in oral and injectable formulations, Pharm. Res., 2004, 21, 201-230.
- S. M. Shah, A. S. Jain, R. Kaushik, M. S. Nagarsenker, and M. J. Nerurkar, Preclinical formulations: Insight, strategies, and practical considerations, AAPSSciTech, 2014, 15, 1307-1322.
- W. Dai, L. C. Donga, S. Li, C. Pollock-Dove, J. Chena, P. Mansky, and G. Eichenbaum, Parallel screening approach to identify solubility-enhancing formulations for improved bioavailability of a poorly water-soluble compound using milligram quantities of material, Int. J. Pharm. 2007, 336, 1-11.
- F. Pilotazet, F. Mercier and H. Chibret, Preservative free prostaglandin based ophthalmic solution, US Patent 8,772,337 (July 2014).
- S. Berko, G. Regdon., and I. Eros, Solutol and Cremophor products as new additives in suppository formulation, Drug Dev. Indus. Pharm., 2002, 28, 203-206.
- S. Ali, Kolliphor® HS 15, In Solubility enhancement with BASF Pharma Polymers – Solubilizer compendium, 2011, pp.76-84.
- H. Janet, M. Liz, U.Diana etal., The analysis of Solutol HS 15 using MALDI and ion mobility, In: Proceedings of the 18th International Mass Spectrometry Conference, Bremen, Germany, 2009, PMM–484.
- J. Hammond, L. Meehan, and D. Uria, The analysis of Solutol® HS 15 using MALDI and Ion Mobility - PMM - 484
- H. Zhang, Z. Wang, and O. Liu, Simultaneous determination of Kolliphor® HS 15 and Miglyol® 812 in micro-emulsion formulation by ultrahigh performance liquid chromatography coupled with nano quantity analyte detector (UHPLCNQAD), J. Pharm. Anal., Letters in Drug Design & Discovery, Vol.: 2 (3), 193-195 DOI: 10.2174/1570180053765165
- J. S. Coon, W. Knudson, K. Clodfelter, B. Lu, and R. S. Weinstein, Solutol® HS 15, Nontoxic polyoxyethylene esters of 12-hydroxystearic acid, reverses multidrug resistance, Cancer Res.1991, 51, 897-902.
- V. Bhaskar, A. Middha, S. Tiwari and S. Shivakumar, Identification and reduction of matrix effects caused by Solutol® HS 15 in bioanalysis using liquid chromatography/tandem mass spectrometry, Anal. Bioanal. Tech., 2013, 4, 1-8.
- V. Bhaskar, A. Middha, P. Srivastava, and S. Rajagopal, Liquid chromatography/ tandem mass spectrometry method for quantitative estimation of Solutol® HS 15 and its applications, J. Pharm. Anal. 2014, 121-129.
- G. Thaysen, Application of Solutol® HS 15, Exact, 1999, 3, 7.
- F. Ruchatz, Applications of Solutol® HS 15- a potential solubilizer with a low toxicity, BASF ExAct, 2002, 9, 6-8.
- V. Buhler, Pharmaceutical technology of BASF excipients, June 2008
- D. Yin, C. Chu, X. Ding, J. Gao, H. Zou, and S. Gao, Nonionic amphiphilic surfactant conjuncted polyethyleneimine as a new and highly efficient non-viral gene carrier, Macromol. Res., 2009, 17, 19-25.
- Shubber, D. Vllasaliu, C. Rauch, F. Jordan, L. Illum, S. Stolnik; Mechanism of Mucosal Permeability Enhancement of CriticalSorb® (Solutol® HS 15) Investigated In Vitro in Cell Cultures, Pharm. Res., 2015, 32, 516-527.
- H. Ryoo, C. Park, S. Chi, and E. Park, Development of propofol-loaded microemulsion systems for parenteral delivery, Arch. Pharm. Res., 2005, 28, 1400-1404.
- M. Crew, Bioavailability enhancement- Diffusion of innovation & the adoption of solubilization technologies: Observations of trends & catalysts, Drug Dev. & Del., June 2014.
- K. Patel, V. Sarma and P. Vavia, Design and evaluation of Lumefantrine- oleic acid self nanoemulsifying ionic complex for enhanced dissolution, · Daru-Journal of Faculty of Pharmacy, 2013, 21, 27; DOI: 10.1186/2008-2231-21-27
- P. Umapathi, J. Ayyappan, and S. D. Quine, Development and Validation of a Dissolution Test Method for Artemether and Lumefantrine in Tablets, Trop J Pharm Res 2011, 10, 643–653.
- C. J. H. Porter, N. L. Trevaskis and W. N. Charman, Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs, Nature Reviews, 2007, 6, 230-248.
- Li Liu, Kai Mao, Wenting Wang, Hongchun Pan, Fen Wang, Min Yang, Hong Liu, Kolliphor® HS 15 Micelles for the Delivery of Coenzyme Q10: Preparation, Characterization, and Stability, AAPS PharmSciTech, pp 1-10, 04 September 2015.
- K. Sapra, A. Sapra, S. K. Singh, and S. Kakkar, Self emulsifying drug delivery system: A Tool in solubility enhancement of poorly soluble drugs, Indo Global J. Pharm. Sci, 2012, 2, 313-332.
- N. Sadurni, C. Solnas, N. Azemar and M. J. Garcia-Selma, Study on formation of O/W emulsions, by low energy emulsion method, suitable for pharmaceutical applications, Eur. J. Pharm. Sci., 2005, 26, 438-441.
- N. Heshmati, X. Cheng, E. Dapat, P. Sassene, G. Eisenbrand, G. Fricker and A. Müllertz, In vitro and in vivo evaluations of the performance of an indirubin derivative, formulated in four different self-emulsifying drug delivery systems, J. Pharm. Pharmacol., 2014, 66, 1567-1575.
- J. Zhou, L. Tan, J. Xie, Z. Lai, Y. Huang, C. Qu, D. Luo, Z. Lin, P. Huang, Z. Su and Y. Xie, Characterization of brusatol self-microemulsifying drug delivery system and its therapeutic effect against dextran sodium sulfate-induced ulcerative colitis in mice, Drug Delivery, 2017, 24, 1667-1679.
- D. M. Benbrook, N. B. Janakiram, V. Chandra, G. Pathuri, V. Madka, N. C. Stratton, C. P. Masamha, C. N. Farnsworth, L. Garcia-Contreras, M. K. Hatipoglu, S. Lighfoot, C. V. Rao,, Development of a dietary formulation of the SHetA2 chemoprevention drug for mice, Invest. New Drugs, 2107, https://doi.org/10.1007/s10637-017-0550-0
- J. U. Menon, P. K. Gulaka, M. A. McKay, S. Geethanath, L. Liu and V. D. Kodibagkar, Dualmodality, dual-functional nanoprobes for cellular and molecular imaging, Theranostics, 2012, 2, 1199-1207.
- A. Tan and C. Prestidge, Improving the performance of lipid formulations: Nanoparticle layers and solid hybrid particles, Am. Pharm. Rev., Dec. 2013.
- (a) A. Abdalla, K. Mader, Preparation and characterization of a self-emulsifying pellet formulation, Eur. J. Pharma. Biopharm. 2007, 66, 220–226. (b) A. Abdalla, S. Klein and K. Mader, A new self-emulsifying drug delivery system (SEDDS) for poorly soluble drugs: Characterization, dissolution, in vitro digestion and incorporation into solid pellets, Eur. J. Pharma. Sci. 2008, 35, 457-464.
- T. H. Hassan, Formulation and evaluation of self-nanoemulsifying tablets for the delivery of poorly water-soluble drugs, PhD Thesis, Martin Luther, Germany, 2015.
- S. G. Gumaste, S. A. Pawlak, D.M. Dalrymple, C. J. Nider, L. D. Trombetta and A. T. M. Serajuddin, Development of solid SEDDS, IV: Effect of adsorbed lipid and surfactant on tableting properties and surface structures of different silicates, Pharm. Res. 2013, 30, 3170-3185.
- B. U. Sri, Y. I. Muzib, D. Bhikshapathi, and R. Sravani, Enhancement of solubility and oral bioavailability of poorly soluble drug valsartan by novel solid self-emulsifying drug delivery system, Int. J. Drug Del., 2015, 7, 13-26.
- K. Cerpnjak, A. Zvonar, F. Vrecer, and M. Gašperlin, Characterization of naproxen-loaded solid SMEDDSs prepared by spray drying: The effect of the polysaccharide carrier and naproxen concentration, Int. J. Pharm. 2015, 485, 215-228.
- F. Li, S. Song, Y. Guo, Q. Zhao, X. Zhang, W. Pan and X. Yang, Preparation and pharmacokinetics evaluation of oral self-emulsifying system for poorly water-soluble drug Lornoxicam, Drug Delivery, 2017, Drug Delivery, 2015, 22, 487-498.
- D. V. Ramani and K.P.R. Chowdary, Enhancement of solubility and dissolution rate of efavirenz employing beta-cyclodextrin, Solutol® HS 15 and PVP K30: A factorial study, Int. J. of Pharm. Pharma. Sci., 2012, 4, 491-493.
- J. Varshosaz, V. Hajhashemi, and S. Soltanzadeh, Lipid nanocapsule-based gels for enhancement of transdermal delivery of ketorolac tromethamine, J. Drug Delivery, doi:10.1155/2011/571272
- R. C. Bravo Gonzalez, J. Huwyler, I. Walter, R. Mountfield, and B. Bittner, Improved oral bioavailability of cyclosporin A in male Wistar rats: Comparison of a Solutol® HS 15 containing self-dispersing formulation and a microsuspension, Int. J. Pharm. 2002, 245, 143-151.
- S. Cui, S. Nie, Li Li, C. Wang, and W. Pan, J. Sun, Preparation and Evaluation of Self-Microemulsifying drug delivery system containing vinpocetine, Drug Dev. Indust. Pharm., 2009, 35, 603-611.
- S. Bhattacharyya and R. P. Vuppalapati, Formulation and evaluation of lorazepam microemulsion for parenteral delivery system, Studia Universitatis “Vasile Goldiş”, Seria Ştiinţele Vieţii, 2014, 24, 289-292
- S. Chauvet, A. Barras, R. Boukherrou, and A. Bouron, Lipid nanocapsules containing the non-ionic surfactant Solutol HS 15 inhibit the transport of calcium through hyperforinactivated channels in neuronal cells, Neuropharmacl., 2015, 99, 726-734.
- E. Garcion, A. Lamprecht, B. Heurtault, A. Paillard, A. Aubert-Pouessel, B. Denizot, P. Menei, and J. Benoıt, A new generation of anticancer, drug-loaded, colloidal vectors reverses multidrug resistance in glioma and reduces tumor progression in rats, Mol. Cancer Ther. 2006, 5, 1710 -1722
- C. Dulieu and D. Bazile, Influence of lipid nanocapsules composition on their pptness to freeze-drying, Pharm. Res., 2005, 22, 285-292.
- S. B. Lim, A. Banerjee, and H. Önyükse, Improvement of drug safety by the use of lipidbased nanocarriers, J. Contr. Rel., 2012, 6, 34-45.
- M. Pitorre, G. Bastiat, E. M. Chatel and J. P. Benoit, Passive and specific targeting of lymph nodes: the influence of the administration route, Eur. J. Nanomed. 2015, 2, 121-128.
- A. S. Narang, R-K. Chang, and M. A. Hussain, Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems, J. Pharm. Sci., 2013; DOI 10.1002/jps.23691
- S. Peltier, J-M. Oger, F. Lagarce, W. Couet, and J-P. Benoıt, Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded lipid nanocapsules, Pharm. Res. 2006, DOI: 10.1007/s11095-006-0022-2.
- P. Gao, B. D. Rush, W. P. Pfund, T. Huang, J. M. Bauer, W. Morozowich, M. S. Kuo, and M. J. Hageman, Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J. Pharm. Sci. 2003, 92:2386-2398
- S. Yang, R. N. Gursoy, G. Lambert, and S. Benita. Enhanced oral absorption of paclitaxel in a novel self-microemulsifying drug delivery system with or without concomitant use of P glycoprotein inhibitors. Pharm. Res. 2004, 21, 261-270.
- C. L. Dora, J-L. Putaux, I. Pignot-Paintrand, F. Dubreuil, V. Soldi, R. Borsali and E. Lemos-Senna, Physicochemical and morphological characterizations of glyceryl tristearate/castor oil nanocarriers prepared by the solvent diffusion method, J. Braz. Chem. Soc., 2012, 0, 1-10.
- C. L. Dora, L. F. C. Silva, M. P. Tagliari, M. A. S. Silva, and E. Lemos-Senna, Formulation study of quercetin-loaded lipid-based nanocarriers obtained by hut solvent diffusion method, Lat. Am. J. Pharm., 2011, 30, 289-295.
- M. Joshi, S. Pathak, S. Sharma, and V. Patravale, Design and in vivo pharmacodynamics evaluation of nanostructured lipid carriers for parenteral delivery of artemether: Nanoject, Int. J. Pharm., 2008, 364, 119-126.
- R. H. Müller, M. Radtk, and S. A. Wissing, Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv. Drug Del. Rev., 2002; 54: S131–S155.
- S. Gupta, S. P. Moulik, S. Lala, M. K. Basu, S. K. Sanyal, S. Datta, Designing and testing of an effective oil-in-water microemulsion drug delivery system for in vivo application. Drug Del., 2005, 12, 267–273.
- H. M. El-Laithy, Y. E. Hamza, and S. M. Kandi, Design and hepatoprotective evaluation of biphenyl dimethyl dicarboxylate (DDB) and silymarin solid dispersion and self-micro emulsifying drug delivery systems, Life Science J. 2011, 8, 298-309.
- J. Breitenbach, Soliqs Symposium on Pharmaceutical Melt Extrusion, 2008, October 9–10.
- B. Egger-Heigold, The effect of excipients on pharmacokinetic parameters of parenteral drugs. PhD thesis, Basel University, Sept. 2005.
- K. J. Scheller, S. J. Williams, A. J. Lawrence, B. Jarrott, E. Djouma, An improved method to prepare an injectable microemulsion of the galanin-receptor 3 selective antagonist, SNAP 37889, using Kolliphor® HS 15, Methods X, 2014,1, 212-216.
- L. Gan, Y. Gan, C. Zhu, X. Zhang, J. Zhu, Novel microemulsion in situ electrolyte-triggered gelling system for ophthalmic delivery of lipophilic cyclosporine A: in vitro and in vivo results, Int. J. Pharm. 2009, 365,143–14 9.
- X. Zhao, D. Chen, P. Gao, P. Ding, and K. Li, Synthesis of ibuprofen fugenol ester and its microemulsion formulation, Chem. Pharm. Bull., 2005, 53, 1246-1250.
- Feng L. and Dexi L. L., Circulating emulsions (oil-in-water) as carriers for lipophilic drugs, Pharm. Res., 1995, 12, 1060-1064.
- X. Dong and R. J. Mumper, Nanomedicinal strategies to treat multidrug-resistant tumors: current progress, Nanomedicine, 2010, 5, 596-615.
- J. B. Glen and R. James, Anaestehtic composition containing 2,6-diisopropylphenol, US Patent 4,452,817 (June 5, 1984)
- A. R. Bell, F. Cochrane, G. N. O’Connor, and J. S. Rowe, Formulations for anesthetic use, US Patent 7,326, 735 (Feb. 5, 2008)
- K. Kim, B. Choi, S. Park, S. Lee, L.V. Christensen, J. Zhou, B. Y. Shin, K. Bae, Steven E. Kern, S. Kang, G. Noh, Pharmacokinetics and pharmacodynamics of propofol microemulsion and lipid emulsion after an intravenous bolus and variable rate infusion, Anesthesiology, 2007,106, 926-934.
- E. H Lee, S.H. Lee, D. Y. Park, K. H. Ki, E. K. Lee, D. H. Lee, G. J. Noh, Physicochemical Properties, pharmacokinetics, and pharmacodynamics of a reformulated microemulsion propofol in rats, Anesthesiology, 2008, 109, 436-447.
- A. A. Date and M. S. Nagarsenker, Design and evaluation of microemulsions for improved parenteral delivery of propofol, AAPS PharmaScitech, 2007, 9, 138-145.
- P. K. Dubey and A. Kumar. Pain on injection of lipid-free propofol and propofol emulsion containing medium chain triglycerides: A comparative study. Anesth. Analg. 2005, 101, 1060-1062.
- T. A. Rodrigues, R. A. Alexandrino, M. E. Kanczuk, J. L. Gozzani, L. A. Mathias, A Comparative Study of Non-Lipid Nanoemulsion of Propofol with Solutol and Propofol Emulsion with Lecithin, Rev Bras Anestesiol, 2012; 62, 325-334.
- J. C. Rittes, G. Cagno, M. V. Perez, and L. A. d-S. T. Mathias, Comparative evaluation of propofol in nanoemulsion with Solutol® HS 15 and soy lecithin for general anesthesia, Rev. Bras. Anestesiol, 2016, 66, 225-230.
- J. Y. Sim, S. H. Lee, D. Y. Park, et al. Pain on injection with micro-emulsion propofol. Br J Clin Pharmacol. 2009; 67:316-25.
- B. Larsen, U. Beerhalter, and A. Biedler et al. – Less pain on injection by a new formulation of propofol? A comparison with propofol LCT. Anaesthesist, 2001, 50, 842-845.
- T. E. Morey, J. H. Modell, D. Shekhawat, T. Grand, D. O. Shah, N. Gravenstein, S. P. McGorray, D. M. Dennis, Preparation and anesthetic properties of propofol microemulsions in rats, Anesthesiology, 2006, 104, 1184-90.
- B. T. Hill, K. Deuchars, L. K. Hosking, V. Ling, and R. D. Whelan, Overexpression of P-glycoprotein in mammalian tumor cell lines after fractionated X irradiation in vitro, J Natl Cancer Inst. 1990, 82, 607-612.
- A. Seelig and G. Gerebtzoff, Enhancement of drug absorption by noncharged detergents through membrane and P-glycoprotein binding, Expert Opinion Drug Metab. Toxicol., 2006, 2, 733-752.
- A. V. Kabanov, E.V. Batrakova, V. Y. Alakhov, Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery, J. Contr. Rel., 2002, 82, 189-212.
- E. V. Batrakova, S. Li, V. Y. Alakhov, D. W. Miller, and A. V. Kabanov, Optimal structure requirements for pluronic block copolymers in modifying P-glycoprotein drug efflux transporter activity in bovine brain microvessel endothelial cells, The J. Phrm. Exper. Therapeut., 2003, 304, 845-854.
- P. P. Constantinides and K. M. Wasan, Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: In vitro/In vivo case studies, J. Pharm. Sci., 2007, 96, 235-247.
- S. W. Wang, J. Monagle, C. McNulty, D. Putnam, H. Chen, Determination of P-glycoprotein Inhibition by excipients and their Combinations using an integrated high-throughput process, J. Pharm. Sci., 2004, 93, 2755-2767.
- J. Hou, J. Wang, E. Sun, L. Yang, H-M. Yan, X-B. Jia and Z-H. Zhang, Preparation and evaluation of icariside II-loaded binary mixed micelles using Solutol HS 15 and Pluronic F127 as carriers, Drug Delivery, 2016, 23, 3248-3256.
- T. R. Kommuru, B. Gurley, M. A. Khan, and I. K. Reddy, Self-emulsifying drug delivery systems (SEDDS) of Coenzyme Q10: formulation development and bioavailability assessment. Int. J. Pharm., 2001, 212, 233–46.
- Y. Zhang and L. Benet, The gut as a barrier to drug absorption: combined role of cytochrome P4503A and P-glycoprotein. Clin. Pharmaco., 2001, 40, 159-168.
- C. M. O’Driscol, Lipid-based formulations for intestinal lymphatic delivery. Eur. J. Pharm. Sci., 2002, 15, 405–415.
- H. W. Lin, I. Saul, V. L. Gresia, J. T. Neumann, K. R. Dave, and M. A. Perez-Pinzon, Fatty Acid Methyl esters and Solutol® HS 15 confer neuroprotection after focal and global cerebral ischemia, Transl. Stroke Res., 2014, 5, 109-117.
- M. Bartels, A. M. Vilsendorf, W. T. Kassahun, B. von Gerstenbergk, K. Engelhart, E. M. Vilsendorf, S. Faber, and H. K. Biesalski, Protective effect of antioxidative vitamins against lipid peroxidation in liver ischemia and reperfusion - an animal experimental study, EXCLI Journal 2007, 6, 152-166.
- M. Bartels, H. K. Biesalski, K. Engelhart, G. Sendlhofer, P. Rehak, and E. Nagel, Pilot study on the effect of ischemia and reperfusion induced liver injury by vitamin E: a double blind, randomized, placebo-controlled trial. Clin. Nutr. 2004, 23,1360-70
- L. E. Buckingham, M. Balasubramanian, R. M. Emanuele, K. E. Clodfelter, and J. C. Coon, Comparison of Solutol® HS 15, Cremophor® EL and novel ethoxylated fatty acid surfactants as multidrug resistance modification agents, Int. J. cancer, 1995, 62, 436-442.
- K. Woodburn, E. Sykes, and D. Kessel, Interactions of Solutol HS 15 ad Cremophor EL with plasma lipoproteins, Int. J. Biochem. Cell Biol., 1995, 27, 693-699.
- D. B. Lewis, Current FDA perspective on leachable impurities in parenteral and ophthalmic drug products, AAPS Workshop on Pharmaceutical Stability – Scientific and Regulatory Considerations for Global Drug Development and Commercialization, October 22-23, 2011, Washington, DC
- J. K. Cheung, M. Sharma, A. Dabbara and J. Petersen, The effect of formulation excipients on leachables for IV-administered products, Pharmaceutical Outsourcing, Nov. 30, 2012.
- W. Hu, C. Berdugo and J. J. Chalmers, The potential of hydrodynamic damage to animal cells of industrial relevance: current understanding, Cytotech, 2011, 63, 445-460
- FDA Inactive ingredient database (2018)- listed as PEG 15 hydroxystearates
- Solutol® HS15- Non-clinical safety and toxicology report (BASF)
- K. Kolter and F. Ruchatz, Solutol® HS 15 – Practical aspects for the production of solubilizates, BASF ExAct 2003, 11, 8-9
- I. M. Kapetanovic, M. Muzzio, S. C. Hu, J. A. Crowell, R. A. Rajewski, J. L. Haslam, L. Jong, D. L. McCormick, Pharmacokinetics and enhanced bioavailability of candidate cancer preventative agent, SR13668 in dogs and monkeys, Cancer Chemoth. Pharmacol. 2010, 65, 1109–1116.
- J. M. Reid, C. A. Walden, R. Qin, K. L. A. Ziegler, J. L. Haslam, R. A. Rajewski, R. Warndahl, C. L. Fitting, D. Boring, E. Szabo, J. Crowell, M. Perloff, L. Jong, B. A. Bauer, S. J. Mandrekar, M. M. Ames, and P.J. Limburg, Phase 0 Clinical Chemoprevention Trial of the Akt Inhibitor SR13668, Cancer Prev. Res., 2011, 4, 347-353.
- W. Dulin, Regulatory approval process for drug products containing new excipients with case studies, AAPS 2010, New Orleans, LA.
- R. Velagaleti and S. Ku, Solutol® HS 15 as a novel excipient identification of the need for and implementation of a US regulatory strategy, Pharm. Tech., Nov. 2010, 108-110.
- A. H. Stokes, D. C. Kemp, B. Faiola1, H. L. Jordan, C. L. Merrill, J. R. Hailey, R. E. Brown, and D. W. Bailey, Effects of Solutol® (Kolliphor®) and Cremophor® in Polyethylene Glycol 400 Vehicle Formulations in Sprague-Dawley Rats and Beagle Dogs, Int. J. Tox., 2013 32, 189-197.