Abstract
Lipid-based drug products present complications for bacterial endotoxins testing because the Limulus-based detection system functions only in an aqueous environment. By taking advantage of the amphipathic nature of lipopolysaccharides, an oil-water extraction process may be used to separate endotoxins from oily drug formulations. This article presents the results of an endotoxin spiking study that demonstrates the effectiveness of this method, with consideration of the testing parameters that can affect endotoxin recovery.
Introduction
Bacterial Endotoxins Testing (BET) is critically important for demonstrating the safety and purity of parenteral drug products. The test detects and quantifies endotoxins from Gram-negative bacteria using Limulus Amoebocyte Lysate (LAL), an enzymatic detection system derived from the horseshoe crab, Limulus polyphemus.1 Like all biological systems, the LAL enzyme complex has evolved to operate in an aqueous environment. This presents technical challenges for BET when the test article to be examined is insoluble in water.2 Insolubility of the test article can be considered a form of physical interference, which prevents the LAL enzymes from making contact with endotoxins in the sample.
Endotoxins consist chiefly of lipopolysaccharide (LPS), a component of the outer plasma membrane in Gram-negative bacteria. LPS is an amphipathic molecule, consisting of polar saccharide chains and a polysaccharide core, attached to a highly-conserved lipid component known as lipid A.3 Due to its amphipathic nature, LPS forms micelles in every solvent, both aqueous and organic, and its lipid A component allows it to bind to nonpolar materials.4
Because the polysaccharide chains comprise the majority of the chemical structure of LPS, endotoxins are more soluble in water than in nonpolar solvents. It is therefore possible to extract endotoxins from nonpolar liquids by means of liquid-liquid extraction. It has been demonstrated that endotoxins spiked into ultrapure paraffin oil could be extracted into an equal volume of water, with recoveries of 94.2% to 111%.2 BET analysis of nonpolar test articles is therefore possible, provided that the endotoxins can be extracted efficiently into an aqueous solution.
We set out to determine the feasibility of performing BET on a lipid-based drug product and placebo, using liquid-liquid extraction to separate endotoxins from the test articles.
Method Feasibility
The product under examination is a sterile solution of active drug substance in triglycerides, currently in Phase 1 clinical trial. A method was desired that could be used to perform BET on both the active drug product and the placebo, which consists only of the triglyceride vehicle. In order to conserve sample material, initial feasibility testing was performed using unflavored, fractionated coconut oil as a mock sample.
USP <85> states that when samples are subjected to a treatment in order to overcome interference, standard endotoxin must be added to a sample prior to treatment in order to demonstrate that the interference can be overcome without loss of endotoxins.1 In order to be considered free of interference, the measured concentration of endotoxin must be within 50-200% of the known added endotoxin concentration, after subtraction of any endotoxin detected in the solution without added endotoxin. However, because control standard endotoxin (CSE) is typically reconstituted in water, this presented a problem for the validity of such a study: if the added endotoxin remained associated with the water in which it had been reconstituted, then a true extraction event would not have taken place; the endotoxin would not have been in the oil to begin with.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
This technical obstacle was overcome by reconstituting CSE in dimethyl sulfoxide (DMSO), as described by Schlösser.5 DMSO is a polar aprotic solvent that is fully miscible in both oil and water. A vial containing 500 ng of CSE was reconstituted with 5.0 mL DMSO, yielding a solution of 100 ng CSE/mL. 10.0 μL of this CSE solution was then added to 990 μL of oil, for a final endotoxin concentration of 1 ng CSE/mL.
The pre-extraction endotoxin content of the CSE/oil solutions, in endotoxin units per mL (EU/mL), could not be directly determined, because the activity of this 500 ng/vial CSE had not been quantified with the turbidimetric LAL that would be used in the assay. To account for this, an extraction control solution was prepared by adding 10.0 μL of CSE solution to 990 μL of lysate reagent water (LRW). The endotoxin activity of the oil samples was therefore assessed in a relative manner, as a percentage of the activity observed in the extraction control (% extraction efficiency). A sample negative control, to which no endotoxin had been added, was also subjected to the extraction procedure. To ensure homogeneity, all samples were vortexed for 30 seconds prior to use.
To perform the extraction, 0.5 mL of each sample replicate was added to 4.5 mL LRW in separate 15-mL, pyrogen-free centrifuge tubes; this constituted a 1:10 sample dilution. The four CSE/oil tubes were capped and vortexed for 5, 15, 30, and 50 minutes. The water sample (extraction control) and negative control were both vortexed for 50 minutes. After vortexing, the oil samples showed an extensive emulsion at the lipidaqueous boundary. This emulsion did not disperse after one hour of rest time, so all tubes were centrifuged for 10 minutes at relative centrifugal force (RCF) = 2900 g, which forced the complete separation of the lipid and aqueous layers.
Initially, vortexing was performed with depyrogenated glass beads to ensure thorough homogenization; however, this resulted in the formation of a solid white precipitate upon vortexing. It was concluded that this solid matter was produced by friction between the glass beads and the walls of the centrifuge tube. Subsequent attempts omitted the beads, and no further precipitate formation was observed.
For each sample, 200 μL of the aqueous phase was withdrawn from the tube and added to 1.8 mL LRW for a 1:100 sample dilution, which was vortexed for 30 seconds prior to use. All 1:100 sample dilutions were tested via the USP <85> kinetic turbidimetric technique, using four unspiked sample wells and four wells spiked to contain 0.5 EU/mL as a positive product control (PPC). The purpose of the PPC was to demonstrate that no interfering substances from the original test article remained in the 1:100 test solution. Per USP <85>, the accepted range for PPC recovery is 50-200%. Testing also was attempted at the 1:10 dilution, but small amounts of residual oil were transferred to the microplate in each sample well, and PPC recovery was low (43-65%), so further testing at this dilution was discontinued.
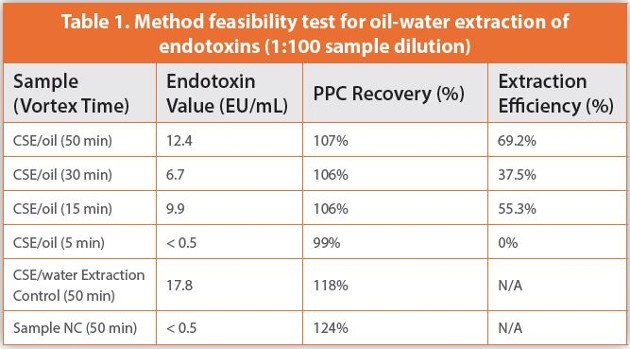
The initial feasibility test demonstrated that endotoxins could be recovered from an oily test article (Table 1). The 1:100 sample dilution did not interfere with the assay, as evidenced by PPC recovery values near 100% for all samples. Extraction efficiency was acceptable with 50 minutes of vortexing, but was markedly lower with only 30 minutes of extraction, and fell to undetectable levels with only 5 minutes of extraction.
Feasibility testing also demonstrated that the endotoxin activity of CSE reconstituted in DMSO declined sharply over time, from 25.1 EU/mL on the day of reconstitution to 17.8 EU/mL after 1 day of storage at 2–8°C. (DMSO has a freezing point of 19°C, so CSE was vortexed for 5 minutes to resuspend after thawing.) Because of this, subsequent testing used CSE in DMSO that had been freshly prepared on the day of use.
Product-Specific Method Verification
Method verification for drug product and placebo formulations proceeded along the same general approach described for method feasibility. For each formulation, the oil-water extraction was performed on an unspiked sample (sample A), a sample spiked with CSE in DMSO to contain 1 ng CSE/mL (sample B), and an equivalent volume of LRW spiked to contain 1 ng CSE/mL (sample C). To improve the accuracy of the CSE spike, the volume was increased to 50.0 μL of CSE in DMSO, added to 4.95 mL of sample.
As in the feasibility test, 0.5 mL of sample was added to 4.5 mL LRW for a 1:10 dilution, vortexed at high speed for 50 minutes, then centrifuged at RCF = 2900 g for 10 minutes to separate the oil and water phases. After centrifugation, the lipid phase of each sample A and sample B was drawn off with an automatic pipette, and the aqueous phase of all samples was vortexed 30 seconds to re-homogenize the endotoxin.
A test dilution of 1:100 was prepared from the aqueous phase, as described for the feasibility test. Additionally, a higher “pass point” dilution of 1:400 was prepared from the 1:100 dilution, in order to establish a validated range of dilutions at which the method could be performed. The use of higher dilutions of 1:800 and 1:1000 was attempted, but results were inconsistent at these dilutions because of high variability in the amount of endotoxin recovered from samples B and C, and these eff orts were discontinued. Dilutions higher than 1:1000 were not tested, because the estimated activity of the spiked CSE in DMSO would fall below the limit of detection (0.005 EU/mL).
All samples were tested by the USP <85> kinetic turbidimetric technique. Equivalent dilutions of samples A, B, and C were tested on the same microplate to ensure comparability of results. The test was performed independently by three analysts to demonstrate intermediate precision.
All analysts obtained extraction effi ciencies in the range of 50-200% required by USP <85> (Table 2). The measured endotoxin activity of the spiked water control (sample C) was uniformly lower than in the feasibility test, ranging from 5.7 to 8.3 EU/mL; this is attributable to the use of a new lot of LAL reagent during the method verification. Endotoxin content in the unspiked sample (sample A) was below the limit of detection for all analysts.
None of the tested sample dilutions exhibited interference with the positive product controls. Across all dilutions and trials, PPC recovery ranged from 100% to 138% for drug product, and from 101% to 127% for placebo.
Conclusion
The results of the method verification demonstrate that endotoxins can be successfully recovered from a lipid sample matrix by liquidliquid extraction. CSE reconstituted in DMSO can be effectively and homogeneously dissolved into lipid samples, and this spiked endotoxin migrates efficiently into the aqueous phase when diluted 1:10 with water. This extraction process can be performed simply by vortexing at high speeds, but time is a critical factor; consistent recovery was not observed for vortex times below 50 minutes.
This study highlights a number of additional technical challenges that must be considered when validating the endotoxin extraction procedure for a new product. Unlike natural endotoxins, CSE settles out of solution over time, so the lipid and aqueous layers must be separated after extraction so that CSE can be resuspended. Emulsion of the layers can be quickly overcome through centrifugation, provided that the aqueous layer is then vortexed to re-homogenize the CSE.
After extraction, direct testing of the aqueous layer proved inadvisable, as enough residual oil remained to interfere with the assay. A ten-fold dilution from this layer, for a 1:100 overall dilution, proved effective in preventing this interference; however, subsequent serial dilutions introduced wide variability in results. The most likely explanation for this is that the CSE adhered to the pipettes that were used in these subsequent dilution steps. This suggests that studies with spiked high-potency CSE should involve as few serial dilutions as possible. Any dilutions that are necessary should be performed in borosilicate glass pipettes, rather than with automatic pipettors, because the lipid A portion of the LPS molecule has a high affinity for nonpolar plastic pipette tips.
USP <85> allows for samples to be tested at dilutions up to the maximum valid dilution (MVD), which is a function of the endotoxin limit for the test article in question. When validating an extraction method such as this one, however, the dilution range is further constrained by the need to quantify the recovered endotoxin. Different lots of CSE vary in their activity, and different lots of LAL vary in their responsiveness to different lots of CSE; and even when CSE activity has been quantified for a given lot pairing, it is done with CSE reconstituted in water, not in DMSO. Because of all these factors, the exact activity of endotoxin being spiked into a sample will not be known until the test is run. The use of an extraction control, in which an equal amount of CSE is spiked into water and tested alongside the lipid samples, ensures a more accurate determination of extraction effi ciency than assuming a theoretical value of EU/mL that has been introduced into the sample. A maximum dilution can then be determined based on the level of endotoxin activity observed.
References
- USP <85> Bacterial Endotoxins Test. In: United States Pharmacopeia and National Formulary, USP42-NF37. Rockville, MD: United States Pharmacopeial Convention; 2019:6434.
- Chen D. A New Method for the Analysis of Bacterial Endotoxins in Ultrapure Paraffi n Oil. J of Analytical Methods in Chemistry. 2014; Article ID 575246, 4 pages.
- Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8(2):217-25.
- Millipore Sigma. Lipopolysaccharides technical document. Available at https://www.sigmaaldrich.com/technical-documents/protocols/biology/lipopolysaccharides.html. Accessed October 8, 2019.
- Schlösser, A. “Endotoxin testing of oily substances.” ECA Conference, 2009, Berlin, Germany. Unpublished conference presentation, 2009.
Author Biographies
Christopher Lester is a senior scientist specializing in bacterial endotoxins testing within microbiology testing services at the PPD® Laboratories GMP lab in Middleton, Wisconsin. Chris and his team perform bacterial endotoxins testing, which includes method development, verification and stability testing for a wide variety of drug formulations. Chris has been performing GxP testing in the contract research industry since 2014.
Timothy Ramsey is the lab manager of microbiology testing services at the PPD® Laboratories GMP lab in Middleton. Tim has more than 20 years of experience in managing microbiology operations in contract laboratory and pharmaceutical manufacturing environments.
Jonathon Salsbury is the associate director of microbiology and physical testing services at the PPD® Laboratories GMP lab in Middleton. Jonathon has been involved with pharmaceutical sciences for more than 20 years, including over 15 years in the contract research industry.