Introduction
Drug development pipelines are inundated with candidates with poor aqueous solubility. To achieve target exposure and be efficacious, these compounds often require use of a bioavailability-enabling technology that enhances solubility and/or dissolution rate. Drug development scientists have many such technologies at their disposal, but selection of the optimal technology remains crucial to ensure success at reasonable cost and timeline. The ultimate goal in any drug development program is to propose the simplest, most cost-effective solution for developing the targeted drug product, and the choice hinges on applying an integrated approach where bioperformance, stability, and manufacturability are considered throughout the development cycle.
Within this context, amorphous solid dispersions (ASDs) have emerged as the technology of choice for improving bioavailability, driven mainly by the technology’s applicability across a diverse compound property space and its scalable manufacturing processes, offering the flexibility and control needed for an optimized drug product.1,2
There are three key stages of development to move bioavailability-enabled formulations, based on ASDs or other enabling technologies, from concept to product: (1) technology selection, (2) formulation screening, and (3) dosage form development. The principles and approach for developing ASD drug products, which are generally applicable to other technologies, are critical for meeting target product profiles and development timelines.
Technology Selection
The timing, cost, and success of a development program for a bioavailability-enabled formulation hinges on selecting the right technology for the compound under development, keeping in mind (1) the physicochemical characteristics of the compound, (2) technology applicability, and (3) the target characteristics and performance attributes of the final drug product based on the rate-limiting step to absorption and target product profile. Generally, technology selection starts with measuring (or calculating) compound physicochemical properties - e.g., lipophilicity (logP), melting temperature (Tm), glass-transition temperature (Tg), permeability, and aqueous solubility. This information is used to identify potential barriers to absorption that may dictate which technology may be optimal. Barriers to absorption generally include drug dissolution rate, solubility, and/or permeability, all of which play a direct role in drug absorption.
Compound guidance maps, generated from large historical data sets of poorly soluble compounds advanced through the various enabling technologies, are then used for initial technology assessment to identify which technology is best suited for the compound, given its physicochemical properties and the problem statement being addressed. Figure 1 shows two guidance maps that formulators have developed to make initial assessments for compounds that require a bioavailability-enhancing formulation. They are best used with industry-wide compound classification systems - such as the Biopharmaceutical Classification System (BCS) and Fraction Absorbed Classification System (FaCS) - and are designed to identify barriers to absorption based upon compound properties and dose.3,4 These maps act as an exercise to set “goal posts” at the beginning of a development program, identifying viable technologies for accomplishing the program goals. They can also be used as aids in overcoming challenges encountered during later stages of development, e.g., during manufacture of the drug product intermediate and final dosage form, which can be technology-specific. The applicability of ASD technology in each map encompasses the compound property landscape that many common BCS Class II/IV candidates tend to occupy (aqueous solubility < 100 μg/mL, logP = 3 to 6, Tm > 100°C).5
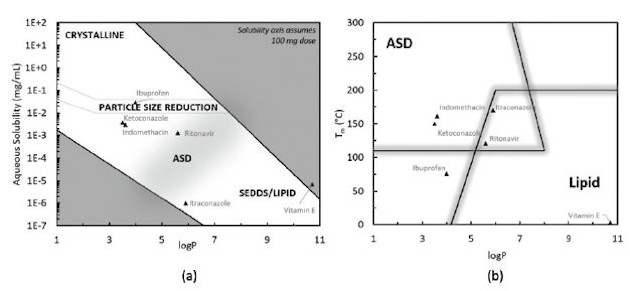
Figure 1. Maps for guiding technology selection for compounds with low aqueous solubility, showing logP (i.e., lipophilicity) versus aqueous solubility, assuming a 100-mg in vivo dose (a); and logP versus Tm (b). Figures adapted from Williams et al.,6 SEDDS = self-emulsifying drug delivery system.
The final step in technology selection involves consideration of the target characteristics and performance attributes of the final drug product. This involves consideration (and clear definition) of the problem statement, development path and critical performance attributes (CPAs) of the final product, in vivo dose, dosing frequency, and target species and population should all play a role in the final decision on which technology to pursue because they will impact product design. Finally, attention to technology precedence is important. It not only provides initial clarity, but helps ensure a clear development path to a final drug product if the technology has been previously used in the clinic or, preferably, in an approved commercial product. Fortunately, many bioavailability-enabling technologies inclusive of particle size reduction, lipid-based and solid amorphous, e.g., ASDs, have a proven track record and have been approved for use in commercial products. 6-9
Formulation Screening
Formulation screening for ASDs is focused on three main considerations: (1) performance, (2) stability, and (3) manufacturability, again keeping the ultimate product goals in mind.
Performance
ASDs are comprised of amorphous drug molecularly dispersed within an excipient matrix, typically polymer, targeted to provide improved aqueous solubility and thermodynamic driving force for absorption over their lower energy crystalline forms. For ASD formulations, initial screening typically involves measuring the compound’s crystalline and amorphous solubility in various dissolution media to assess the overall maximum solubility benefit achievable with the amorphous form. Polymer screening is commonly coupled with these measurements to determine which polymer - when predissolved in dissolution medium - best sustains supersaturated concentrations of drug or, ideally, prevents precipitation to a lower concentration.
Once these measurements have been made, dispersion polymers and drug loadings are selected for initial ASD manufacture and evaluated using biorelevant dissolution testing.10,11 Test selection is critical to the design and evaluation of the formulation, ensuring the CPAs identified during technology selection are met – target dissolution rate, amorphous solubility versus pH, or sustainment of supersaturation, to name a few.
There is a wide array of in vitro dissolution tools available for formulation screening that vary in complexity and overall design. Some of these tools, shown in Figure 2, are based on a fiber-optic ultraviolet (UV) detection platform for rapid and real-time data collection.
At early formulation screening stages, information obtained from these in vitro methods is designed to rank formulations after evaluating each in terms of the common CPAs. Lead formulations are selected from these criteria and then evaluated for physical and chemical stability.
Stability
Stability is a key factor in advancing a viable formulation to ensure acceptable long term shelf life (typically, two years) and define manufacturing and packaging requirements. Formulations are tested under accelerated conditions - elevated temperature and humidity at specifications set by the International Conference for Harmonisation (ICH) - in various packaging configurations for specified times. Samples are pulled at defined timepoints and their chemical and physical states are analyzed to determine formulation stability. ASD formulations are evaluated to ensure the ASD shows no evidence of crystallization or chemical degradation of the drug above a certain threshold. Table 1 shows common tools used to test ASD stability.

Figure2. Test methodologies for the characterization of ASD bioperformance
Manufacturability
The most prevalent manufacturing processes for producing ASDs are spray drying and hot melt extrusion (HME). Wyttenbach and Kuentz recently reported that of 20 marketed ASD drug products, 14 (70%) were produced via these processes.12 The main reasons these manufacturing processes are preferred are process scalability, flexibility, global capacity among contract development and manufacturing organizations (CDMOs), and years of technological precedence in the pharmaceutical industry. A few general considerations for ASDs manufactured using each process are highlighted in Table 2 with additional details below.
Spray Drying
Spray drying is a solvent-based process in which a drug and polymer (and other excipients, if necessary) are dissolved in a common solvent or mixed solvent system. The spray solution is fed through an atomizer that forces the solution to break up into small droplets within a cyclone chamber. The droplets are then exposed to a hot drying gas that rapidly evaporates the solvent (in milliseconds) from the droplets, leaving isolated amorphous solid dispersion particles, producing a powder with controllable particle size, density, and flow. The powder is then subjected to a secondary drying process to remove any residual solvent.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
A few key processing variables should be considered when spray drying ASDs: the total solids concentration in the spray solution (defining throughput); process conditions - such as solution viscosity, nozzle selection, inlet and resulting outlet temperatures, drying gas flow rate, to name a few - that dictate ASD particle characteristics such as particle size, density, and morphology (since these may impact downstream processing); and factors that may affect the physical and chemical stability of the formulation during spray drying and secondary drying. A thorough review of spray-drying process development for ASDs has been published by Dobry et al.13
Hot-Melt Extrusion (HME)
HME is a thermal-driven mixing process in which a drug and polymer are fed through an extruder at processing temperatures that cause the drug to either melt into the polymer matrix (when the processing temperature > drug Tm) or dissolve into the polymer matrix (processing temperature < drug Tm). In either case, the process typically produces an extrudate with drug molecularly dispersed within the polymer matrix (with or without other excipients) that can be milled into solid ASD particles.
A key consideration for ASDs that are produced via HME is ensuring the processing temperatures do not degrade the polymer or the drug. Balanced with these considerations is defining conditions that produce an ASD that is a single homogeneous phase, since the quenching rate of HME is much slower than that of spray drying (many seconds versus milliseconds). Defining a suitable processing space requires an understanding of the miscibility of the drug and excipient matrix, because drug loading in the ASD intermediate may need to be limited to achieve a physically stable powder once manufactured. For a detailed review on using HME to produce ASDs, refer to Simões et al.14
Dosage Form Development
Before they can be administered to the patient in an orally bioavailable form, ASD intermediates must be incorporated into oral dosage forms. ASDs are amenable for use in a variety of solid oral dosage forms including tablets, capsules, suspensions and sachets, delivered with immediate and modified release, and this flexibility has facilitated the technology’s rapid adoption. Dosage form selection hinges upon factors such as drug development stage, patient population (e.g., geriatric and pediatric patients may have special needs), and desired market image. Commercial ASDs are most commonly formulated as tablets or capsules, with tablets being the most popular consistent with approval trends and patient preferences.12,15-17
Development of a robust ASD dosage form requires an understanding of the key ASD attributes driving performance, stability, and manufacturability. For example, ASD particles made using spray drying tend to be smaller and more porous than ASD particles made using HME.18-21 As a result, spray-dried ASD particles tend to have superior dissolution rate and tablet compression properties, whereas HME particles tend to have higher bulk and tapped densities and superior flowability during downstream processing.18 To increase density and improve flowability, spray-dried ASD particles are often dry granulated before being compressed into tablets. In contrast, HME extrudate is often milled and then directly compressed into tablets with no dry granulation step.

When developing immediate-release dosage forms, the goal is typically to develop a formulation that achieves equal or better in vitro dissolution performance than the ASD intermediate. Achieving this goal requires careful selection of (1) the type and amount of excipients in the formulation and (2) processing specifications. Common excipients in ASD formulations include brittle and/or ductile fillers, disintegrants, lubricants, and glidants. While most ASDs disintegrate and disperse rapidly from tablets or capsules, there are few instances where undesired gelling due to the presence of the ASD dispersion polymer can result in poor disintegration and dispersal.22 In these instances, minimizing the fraction of ASD in the formulation and/or adding certain types of inorganic salts to the tablet or capsule can decrease gelling and improve drug release.23 If such strategies are used, dosage form burden can become a challenge, since the dosage form size will increase and the number of dosage units required for an efficacious dose may rise. However, these challenges can be mitigated using particle-engineering approaches or through strategic design of dosage-form architecture.21,24
Conclusions
ASD technology has been steadily progressed to become the predominant approach for addressing solubility and bioavailability challenges. However, formulation and development of an ASD drug product requires a deep understanding of bioperformance, stability, and manufacturability given the added complexity in design, scaleup, and stability. Each compound presents its own unique challenges during the development process, but a robust generalized strategy can help mitigate some of these risks and provide a streamlined approach. Several best practices in the areas of technology selection, initial formulation screening, and dosage form manufacture provided a clear pathway for designing, developing, and manufacturing a successful ASD drug product.
References
- Mendonsa N, Almutairy B, Kallakunta VR et al. Manufacturing strategies to develop amorphous solid dispersions: An overview. J Drug Deliv Sci Technol. 2020;55:101459.
- Singh A, Van den Mooter G. Spray drying formulation of amorphous solid dispersions. Adv Drug Deliv Rev. 2016;100: 27-50.
- Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3): 413-420.
- Sugano K, Terada K. Rate- and extent-limiting factors of oral drug absorption: Theory and applications. J Pharm Sci. 2015;104(9):2777-88.
- Dahan A, Miller JM, Amidon GL. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11(4): 740-6.
- Williams HD, Travaskis NL, Charman SA et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65(1): 315.
- Friesen DT, Shanker RM, Crew MD, Smithey DT, Curatolo WJ, Nightingale JAS. Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: An overview. Mol Pharm. 2008;5(6):1003-1019.
- Li M, Azad M, Davé R, Bilgili B. Nanomilling of drugs for bioavailability enhancement: a holistic formulation-process perspective. Pharmaceutics. 2016;8(2): 17.
- Sahbaz Y, Williams HD, Nguyen T-H et al., Transformation of poorly water-soluble drugs into lipophilic ionic liquids enhances oral drug exposure from lipid based formulations. Mol Pharm.2015;12(6): 1980-1991.
- Stewart, A.M., et al., Impact of Drug-Rich Colloids of Itraconazole and HPMCAS on Membrane Flux in vitro and Oral Bioavailability in Rats. Mol Pharm, 2017. 14(7): p. 2437-2449.
- Stewart A, Yates I, Mudie D et al., Mechanistic study of belinostat oral absorption from spray-dried dispersions. J Pharm Sci. 2019;108(1):326-336.
- Wyttenbach N, Kuentz M. Glass-forming ability of compounds in marketed amorphous drug products. Eur J Pharm Biopharm. 2017;112:204-208.
- Dobry DE, Settell DM, Baumann JM, Ray RJ, Graham LJ, Beyerinck RA. A model-based methodology for spray-drying process development. J Pharm Innov. 2009;4(3):133-142.
- Simões FM, Pinto RMA, Simões S. Hot-melt extrusion in the pharmaceutical industry: toward filing a new drug application. Drug Discov Today. 2019;24(9):1749-1768.
- Jermain SV, Brough C, Williams RO. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery – An update. Int J Pharm. 2018; 535(1):379-392.
- Lu E, Li S, Wang Z. Biorelevant test for supersaturable formulation. Asian J Pharm Sci, 2017;12(1):9-20.
- Vasconcelos T, Marques S, das Neves J, Sarmento B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv Drug Deliv Rev. 2016;100: 85-101.
- Iyer R, Hegde S, Zhang Y-E et al. The impact of hot melt extrusion and spray drying on mechanical properties and tableting indices of materials used in pharmaceutical development. J Pharm Sci. 2013;102(10):3604-13.
- Mahmah O, Tabbakh R, Kelly AL, Paradkar A. A comparative study of the effect of spray drying and hot-melt extrusion on the properties of amorphous solid dispersions containing felodipine. J Pharm Pharmacol. 2014;66(2):275-84.
- Boersen N, Lee T W-Y, Shen X, Hui H-W. A preliminary assessment of the impact of hotmelt extrusion on the physico-mechanical properties of a tablet. Drug Dev Ind Pharm. 2014;40(10):1386-1394.
- Ekdahl A, Mudie D, Malewski D, Amidon G, Goodwin A. Effect of spray-dried particle morphology on mechanical and flow properties of felodipine in PVP VA amorphous solid dispersions. J Pharm Sci. 2019;108(11):3657-3666.
- Démuth B, Nagy ZK, Balogh A et al., Downstream processing of polymer-based amorphous solid dispersions to generate tablet formulations. Int. J.Pharm. 2015;486(1): 268-286.
- Hughey JR, Keen JM, Miller DA, Kolter K, Langley N, McGinity JW. The use of inorganic salts to improve the dissolution characteristics of tablets containing Soluplus®-based solid dispersions. Eur J Pharm Sci. 2013;48(4):758-766.
- Mudie DM, Buchanan S, Stewart A et al. A novel architecture for achieving high drug loading in amorphous spray dried dispersion tablets. Int J Pharm: X. 2020;2:100042.