Given the recent successes of recombinant Factor C, there has been an increase in commercial supplier comparison studies for recombinant Factor C (rFC). The results of such studies appear to have been wholly adopted at face value as reflected in recent articles1 and a new USP draft chapter.2 The problem with the adopted thesis is that it is demonstrably false. The underlying thesis is: rFC underestimates endotoxin recovery in contaminated deionized water, therefore, it must not be detecting all the endotoxin and therefore presents a “health risk”. Unpacking all the inaccuracies accepted at face value in this thesis includes the topics listed below (i-iv). The use of natural water that has not been purified (including deionized water) is an inappropriate matrix for comparison as it contains a thick background of organic materials including beta-glucans that give false positive responses with LAL and not with rFC.
(i) One contention is that microorganisms and/or endotoxin from natural waters (including DI) can “bleed through” the purification process to bring patient risk. In reality, the reason stated for using source / DI water is that the study promoters couldn’t find any contaminated purified water (PharmaLab conference, Dusseldorf Germany, Nov. 2019). Does this not speak to the false proposition of “bleed through”? Rather, biofilm is the reason for very occasional contamination of purified waters by Gram Negative bacteria (GNB) and these have been identified as common GNB types.3-5
(ii) Another contention is that the use of beta-glucan blocking buffer will nullify the false positive LAL Factor G pathway. This contention has been repeated in the USP referenced draft chapter with the instruction for the test user to “use blocking buffer”. Historical data disproving this contention exists6 and was experimentally repeated as described here using modern LALs.
(iii) The piling on of irrelevant “kitchen-sink” theories as to why rFC values are lower include everything except the real reason they are lower: LAL false positive reaction to beta-glucans in source waters that is not seen in purified waters.
(iv) Finally, the addition of a “wish list” of rFC requirements in the draft USP chapter <1085.1> goes beyond the current characterization of LAL.
(i) The use of natural water that is used as a source for pharmaceutical water is not pharmaceutical water for injectable drugs. DI water is a so-called “source water” in that it is as received from the source (municipal or well, etc.) except in that it has been deionized (not purified). Such source water will differ at every location. It is neither microbiologically controlled nor does it have total organic carbon (TOC) control or testing. It is certain to contain both glucans and cellulosic residues and therefore promote more than one kind of false positive results in LAL (beta-glucan and LAL-Reactive material, LAL-RM).7-8 Both have been well documented. Glucan blocking buffer does not block all glucans present in nature and does not block the effects of LAL-RM at all (Roslansky and Novitsky). Neumeyer9 makes it clear that to be a “pharmaceutically relevant water”, the TOC test is integral. TOC is a hallmark of purified water system control for injectable drugs.
The proposal that “rFC underestimates endotoxin” is easy to suggest and easy to repeat as based on studies using non-purified water. However, what is specifically being claimed? Is it the claim that all LAL gives the same result (i.e. any LAL gives a higher result than rFC)? Users know this to be intuitively false. LAL results vary sometimes drastically in the presence of glucans, cellulosic or detergent/ zwittergent substances.6 Any rigorous study claiming to definitively demonstrate non-equivalence should at least employ diff erent LAL types including chromogenic and turbidimetric. Moreover, it must take into account the presence of beta-glucans beyond the casual use of beta-glucan blocking buffer by using enzymes in lieu of blocking buffer and confirmation with beta-glucan testing. If some LAL reactive structures from strange microorganisms (Sphingomonas etc.) are LAL reactive due to either Factor G or another factor yet undetermined in LAL, but is not present in purifi ed water where potential contaminant’s origin derive from GNB biofi lm, then what is the relevance to pharmaceutical QC? According to Sandle: “Given that biofi lms tend to contain predominantly Gramnegative bacteria it is unsurprising that there is an association between biofilms and endotoxin.”
A Charles River study11 showed that three odd organisms accounted for almost half of the flora of “pre-treatment water” samples. These included Sphingomonas (23%), Acidovorax (12%), and Caulobacter (9%). Only Sphinomonas was found amongst those listed in Sandle’s 15 year study of purifi ed water system contaminants (1%).4 Sphingomonas doesn’t contain LPS, but they contain glycosphingolipids, specifically ceramide.
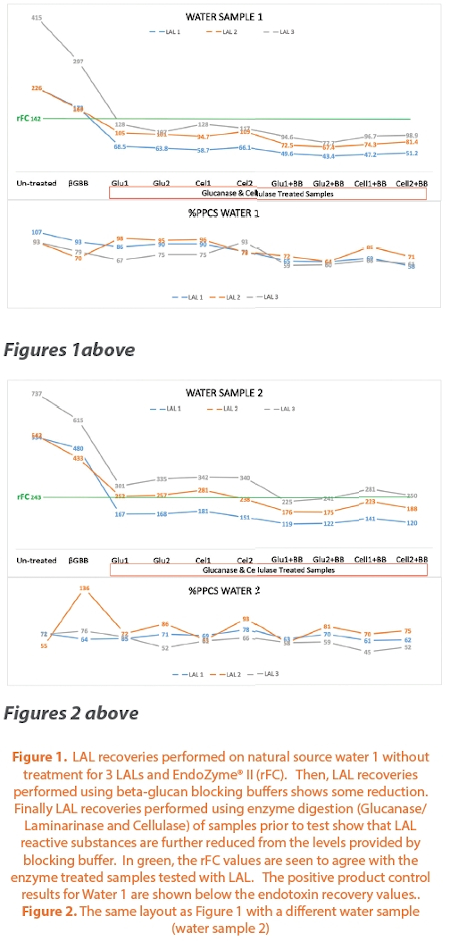
(ii) Beta-glucan blocking buffer is widely believed to be able to block all the eff ects of β-glucans on LAL and thus to ensure that LAL remains the gold standard result for endotoxin in a beta-glucan containing matrix. Data generated using a couple of different natural water sources agrees with the original Roslansky and Novitsky study6 as well as a recent previous study performed with currently available LALs in showing the pattern that LAL recovery is lessened by the use of beta-glucan blocking buffer but signifi cant remaining activity is further reduced by using enzymes that destroy glucans and/or cellulosic residues but that have no effect on endotoxin. See Figure 1-4. The data includes positive product controls (PPC’s) as required to purport that any endotoxin test data is valid. If you see comparison data that has no PPC controls included then that data cannot be claimed to demonstrate any valid comparison. This is because low PPCs indicate inhibition and inhibition will give lower results for LAL or rFC. However, inhibition is routinely overcome via validation where a suitable dilution is determined and the routine test is then only performed without such inhibition or enhancement.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
The enzymes used were prepared and performed as per the Roslansky and Novitsky paper including controls showing (a) the enzymes are not contaminated and (b) the enzymes do not destroy endotoxin. The methods of enzyme treatment should be optimized to achieve these two goals prior to use.
(iii) The piling on of irrelevant “kitchen-sink” theories has picked up steam and is being used to support erroneous conclusions. The “Factor B is needed” theory12 claims that Factor C alone is insufficient to activate the endotoxin reaction. This is false as previously described13 as it is Factor C that acts as a receptor on the amebocyte granulocyte to release the other cascade factors, including Factor B. If Factor B were needed to bind endotoxin then it could not be released from the granulocyte as only Factor C is made by the hepatocytes and located as the receptor on the granulocytes.14-15
(iv) Finally, the addition of a “wish list” of rFC requirements in the proposed USP <1085.1> draft chapter goes well beyond the current requirements for the characterization of LAL. A few of the “new requirements” from the informational only (i.e. not really required) USP draft chapter <1085.1> include the following statements:
- “Formal study of performance characteristics of the analytical methods using recombinant reagents comply with the technical analytical testing requirements of <1225>…”
- “Comparability: Given the complexity of the lysate prepared from disrupted amebocyte granules, comparability of the recombinant reagents to naturally sourced lysate using endotoxins from autochthonous manufacturing sources is of particular importance… equivalency of results per <1223>”
- “choice of species of the horseshoe crab as the source of the recombinant reagents”
- “optimal number of cascade zymogen proteases in the recombinant reagent formulation”
- “the type and control of cell lines used as the expression system”
- “identification and impact of any post-translational modification of the recombinant zymogen proteases”
- “consistent functionality and stability of the chosen recombinant components selected by the reagent manufacturer”
- “It is incumbent on the user of these reagents to assure that the manufacture, distribution, storage, stability and use of the selected recombinant reagent provides sufficient controls to result in consistently comparable results to the naturally sourced LAL reagents…”
- “the designation of the gel-clot limit test as the official referee test applies to all recombinant methods…”
What are test users to make of the sheer weight of the proposed new “informational requirements”? The FDA Q&A Guideline16 of 2012 outlines the appropriate use of USP <1225> for alternative BET validation that has been performed and accepted by FDA for current marketed products tested by rFC. Here the USP draft chapter adds <1223> which is a detailed validation of “microbial enumeration” methods that is often used for microbiological rapid methods.
The document also suggests a list of the following audit controls to be performed by pharmaceutical users desiring to use recombinant reagents. Recombinant reagents are not considered to be drugs, devices, or biological products; therefore, their manufacture may not fully comply with standard drug/device/biological current good manufacturing practices (cGMPs). Table 1 provides suggestions on responsibilities of the supplier and user, which may be helpful as guidelines for auditing.
The referenced Table 1 is a full page spread of proposed requirements to provide before using rFC. The USP Pharmacopeia chapters do not appear to attempt to regulate other test reagents or media such as those used in sterility or bioburden testing to this degree. The USP chapter dictates that “Fluid Thioglycollate Medium” is used, for example in the sterility test, and the temperature it must be incubated at as well as the general media recipe, but they do not require the testing of wild-type or “autochthonous” organisms from diverse sources to ensure growth or dictate how it should be audited by pharmaceutical manufacturers.
The reason LAL has been FDA licensed historically is that it is a bloodborne product.17 Blood-borne accidents such as needle sticks have historically represented a serious risk to medical personnel. The stated intention of such regulation stems from a user safety perspective, not a quality vantage. According to Berzofsky:
One popular misconception is that because endotoxin testing is a final release test for human parenteral drugs, biologics and implantable medical devices, the FDA regulates the test by licensing its manufacturers. But, it’s just not true. Final release testing and FDA licensing do not go hand-in-hand. In fact, endotoxin testing is the only final release test, universally associated with the manufacturing of injectable drugs and medical devices, that is FDA licensed. All other final release tests, i.e. sterility tests, are not.16
The irony in the USP <1085.1> draft chapter proposition of stringent new rFC requirements is that much of the list developed cannot be said to be true for wild-caught horseshoe crab derived reagents.
- For example, “the type and control of cell lines used” has a corollary in nature that is the horseshoe crab genome. A wild-type genome is known to contain variability and mutations (the basis of evolution), organism to organism, over time. Does the LAL manufacturer now confirm the appropriate genetic sequence for each horseshoe crab caught from the sea and bled in the factory? Not at all as this would put them out of business! In fact, this is a good argument in favor of rFC in that these sequences have been cloned and controlled over time in a scientific manner.
- Similarly, the statement: “identification and impact of any post-translational modification of the recombinant zymogen proteases” would require the routine check of the Factor C protein glycosylation pattern while leaving completely unknown the routine configuration of the multiple native LAL proteins as harvested and formulated into various LALs. What is the effect of chloroform extraction on glycosylation of all the LAL proteins? Of course no one knows the detailed molecular contents of LAL.
- Here: “It is incumbent on the user of these reagents to assure that the manufacture, distribution, storage, stability and use of the selected recombinant reagent provides sufficient controls to result in consistently comparable results to the naturally sourced LAL reagents…” USP <1085.1> draft chapter seems to suggest that a comparison study may be needed with each new batch.
The USP <1085.1> draft’s claim that only LAL is “natural” and that rFC doesn’t exist in nature is also debatable (“…recombinant reagents as alternatives to naturally sourced reagents from horseshoe crabs”). A chloroform extracted lysate cannot reasonably be claimed to be “natural”.
- In nature, amebocyte granules are released via Factor C receptor interaction with endotoxin (a discriminating biologic activity) whereas in LAL, cascade reagents are mixed with an anticoagulant and lysed by the osmotic bursting of the granulocytes that releases all the factors to thereby create a protein “soup” (not a natural state). Therefore, LAL employs an “artificial” release and activation of the cascade proteins. Are the proper proportion of enzymes maintained by such a scenario and by different manufacturing practices? This is another advantage of recombinant products where the molecular characterization of a single protein is maintained over time.
- Similarly, the search for “natural endotoxins” seeks to employ freshwater aquatic versions of microbes for comparison whereas the horseshoe crab is exposed to a completely different set of salt-water GNB. Thus, the appeal to what is “natural” and what is “not natural” is always subject to context.
- Where various LALs are used with different means of manufacture (chloroform extraction, addition of zwittergent, both, or as stuck to plastic in a cartridge test) there was no detailed check of the detection of various autochthonous source water contaminants.
The USP <1085.1> draft calls for more characterization for rFC which already highly contrasts the lack of characterization of LAL as a product of hemolymph extraction. USP <1085.1> speaks of the “…complexity of the lysate prepared from disrupted amebocyte granules” as if it is an advantage of LAL whereas, in actuality, the lack of characterization is an inherent weakness of LAL.
- With LAL as a product of chloroform extraction or not (some LAL is and some is not) it is not certain which hemolymph proteins remain in the “soup” and which have been extracted out. Additionally, the remnant of protease inhibitors left denatured by the chloroform extraction treatment may remain in LAL as damaged, non-functional proteins.
- It was many years after LAL acceptance that industry first realized that non-endotoxin reactivity was inherent in LAL via Factor G. The call for the use of a “blocking buffer” by <1085.1> is a tacit admission that LAL cannot get the right answer in the presence of glucans. A “gold standard” reagent that is largely uncharacterized and cannot get the right answer in common circumstances makes for a difficult target in comparison studies using a highly characterized material (rFC).
A thorough understanding of the extensive context surrounding drug testing is needed to restrict the validation of endotoxin to products that will actually be tested routinely (“fit for use”, i.e. purified water, raw materials, container closures and drugs). The reliance on a threatened or endangered animal such as the horseshoe crab for a critical QC function, endotoxin detection, has reached a critical point where global supply chain sustainability requires diversification. Thus, rFC deserves a fair appraisal and reasonable path to commercialization as has been provided by the EU in the publication of Chapter 2.6.32 as adopted as a compendial BET method.
Author Biography
Kevin Williams spent 30 years at Eli Lilly & Company developing endotoxin assays and detection technology in the QC lab. He then worked at Hospira(now Pfizer), Lonza, GE Water before moving to bioMérieux. He has authored several books on endotoxin (Endotoxins second and third edition, Informa) including, most recently Endotoxin Detection and Control in Pharma, Limulus, and Mammalian Systems (2019, Springer Nature).
References
- Functional Challenges for Alternative Bacterial Endotoxins Tests Part 2: Comparability, James Akers, Dennis E. Guilfoyle, David Hussong, Karen McCullough, Robert Mello, Donald Singer, Edward Tidswell, and Radhakrishna Tirumalai, American Pharmaceutical Review, July/August 2020.
- <1085.1.1> USE OF RECOMBINANT REAGENTS IN THE BACTERIAL ENDOTOXINS TEST— PHOTOMETRIC AND FLUOROMETRIC METHODS USING RECOMBINANTLY DERIVED REAGENTS, Pharmacopeial Forum, GENERAL CHAPTERS > GENERAL INFORMATION <1085.1.1> USE OF RECOMBINANT REAGENTS IN THE BACTERIAL ENDOTOXINS TEST - PHOTOMETRIC AND FLUOROMETRIC METHODS USING RECOMBINANTLY DERIVED REAGENTS — PF 46(5).
- Study of Biofilm in Bacteria from Water Pipelines, Mahapatra et al., J Clin Diagn Res. 2015 Mar; 9(3): DC09–DC11.
- Characterizing the microbiota of a water system, T. Sandle, SOJ Microbiology and Infectious Disease, 3(2): 1-8.
- Tim Sandle, The Problem of Biofilms and Pharmaceutical Water Systems, Amer. Pharm. Rev., Friday, December 22, 2017, accessed July 9, 2020.
- Roslansky and Novitsky, Sensitivity of Limulus amebocyte lysate (LAL) to LAL-reactive glucans, Associates of Cape Cod, Inc., Box 224, Woods Hole, Massachusetts 02543, JOURNAL OF CLINICAL MICROBIOLOGY, Nov. 1991, p. 2477-2483.
- Characterization of Limulus Amoebocyte Lysate-Reactive Material from Hollow-Fiber Dialyzers, Pearson et al., APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Dec. 1984, p. 1189-1196.
- Nagasawa et al., Experimental proof of contamination of blood components by (1-3)-β-D-glucan caused by filtration with cellulose filters in the manufacturing process, Jour. Artificial Organs (2003) 6:49–54.
- Process Analytical Technology and Real-Time TOC Testing of Pharmaceutical Grade Water Systems, M. Neumeyer, SUEZ Water Systems, Amer. Pharm. Review, July/August 2020, pg. 114.
- Endotoxin Testing as a Detection Method for Bacterial Biofilms, T. Sandle, Amer. Pharm. Rev., Endotoxin Supplement 2018, pg. 22.
- A global perspective for quantifying all endotoxins within pharmaceutical water systems, N. Reid, PharmaLab Conference presentation, Nov. 2019.
- Innovative mechanism of limulus amebocyte lysate activation to achieve specificity and sensitivity to endotoxin; comparison with recombinant factor c reagents, Masakazu Tsuchiya (Charles River), International Journal of Development Research Vol. 10, Issue, 06, pp. 36751-36756, June, 2020 https://doi.org/10.37118/ijdr.19019.05.2020.
- LAL and rFC Comparison Study Caveats, K. Williams, Amer. Pharm. Rev., American Pharmaceutical Review | July/August 2020, pg. 104.
- Takumi Koshiba, Tomoyuki Hashii, and Shun-ichiro Kawabata, A Structural Perspective on the Interaction between Lipopolysaccharide and Factor C, a Receptor Involved in Recognition of Gram-negative Bacteria, . THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 282, NO. 6, pp. 3962–3967, February 9, 2007.
- Ariki et al., A serine protease zymogen functions as a pattern-recognition receptor for lipopolysaccharides, Proc Natl Acad Sci., 2004, Jan 27;101(4):953-8.
- Guidance for Industry Pyrogen and Endotoxins Testing: Questions and Answers, FDA, 2012, https://www.fda.gov/media/83477/download
- Does Endotoxin Testing = FDA Licensing? By Ronald N. Berzofsky Ph.D., LAL Review, Volume 104, Issue No. 1, published by Cambrex BioScience Inc., Walkersville Maryland, 2004 (Now Lonza).