Authors:
James Akers,1 Dennis E. Guilfoyle,2
David Hussong,3 Karen McCullough,4
Robert Mello,5 Donald Singer,6
Edward Tidswell,7
Radhakrishna Tirumalai8
1Akers Kennedy Associates, Inc., Leawood, KS
2Johnson and Johnson, New Brunswick, NJ
3Eagle Analytical, Houston, TX
4MMI Associates, Whitehouse Station, NJ
5Mello PharmAssociates, LLC, Reisterstown, MD
6Ecolab Life Sciences, St. Paul, MN
7Merck & Co, West Point, PA
8North Potomac, MD
Note: The views expressed in this article are solely those of the individual authors and do not necessarily reflect the opinions of organizations with which the authors may be affiliated.
Introduction
Quality control analyses of all types ensure patient safety by providing evidence that the article under test meets its approved acceptance criteria. These analyses must be validated to demonstrate a capability of detecting, and where possible or required, quantifying the parameter they are purported to measure. Our two prior articles (Akers et al., 2020a; Akers et al., 2020b) have reported what precisely this means for alternative tests in the field of pyrogen and bacterial endotoxins testing. In endotoxins testing any capable alternate method must detect and, depending on the assay, quantify contaminant(s) that are likely to be in the product comparably to the standard compendial method.
As discussed in our second article (Akers et al., 2020b), the concepts of suitability and comparability are not synonymous. Suitability, which for quantitative BET assays is the same “spike/recovery” exercise as the Positive Product Control (PPC), assures that there is no overt interference in the recovery of the calibration standard (RSE or CSE) deliberately added to the product formulation. Suitability is not a contamination recovery determination; it’s a product and assay-specific systems control. Comparability, on the other hand, is irrefutable evidence that actual test results, meaning in the specific case of endotoxins analysis, the measurement of autochthonous endotoxins activity using the candidate assay, are shown to be statistically equivalent or non-inferior to the test result obtained on the same sample using the existing compendial method. Any failure in demonstrating, by appropriately defined qualification studies, that such contaminants can be detected and quantified represents a risk to patient well-being must be thoroughly evaluated.
In this installment, we further clarify the definition and origins of autochthonous endotoxins and provide reported and documented incidents of autochthonous endotoxins found in pharmaceutical and medical device manufacturing operations. We also discuss the concept of a universal model endotoxin system.
Endotoxins From Bacteria Autochthonous to Manufacturing Operations
The term autochthonous has been used in microbiology for decades to describe those microorganisms that are indigenous to, or inhabitants of a specific environment or niche.
These microorganisms exist by adapting their physiologies to their environments, permitting proliferation or simply survival in those unique habitats.
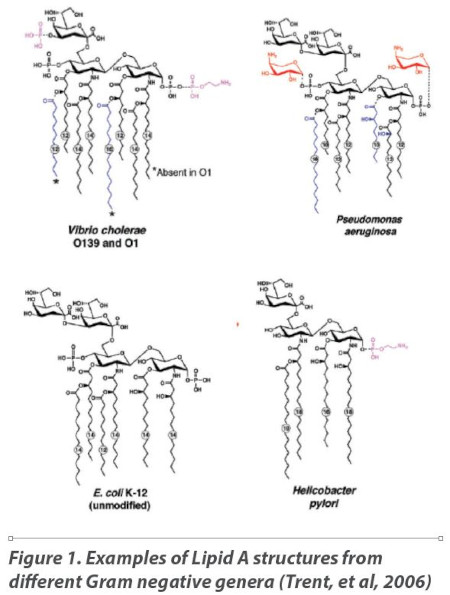
In the four decades since the introduction of the LAL bacterial endotoxins test, research has confirmed that it is the Lipid A moiety of the lipopolysaccharide molecule found in the outer cell membrane that is recognized by both mammalian innate immunity systems and the LAL reagent. Studies on the fine structure of the Lipid A moiety produced by various species of Gram negative bacteria have demonstrated that the Lipid A structure can differ markedly between isolates both in the number and length of acyl (fatty acid) chains depending on the genus, species, and growth conditions. Figure 1 shows the structures of Vibrio cholerae (8 acyl groups) Pseudomonas aeruginosa (7 acyl groups), E. coli (6 acyl groups) and Helicobacter pylori (4 acyl groups) (Trent et al., 2006).
E. coli, the organism identified during initial LAL test development as “typical” of all Gram negative organisms, contains six acyl chains composed of 12-14 carbons each, but as shown in Figure 1, the Lipid A structure can vary considerably across the universe of Gram negative organisms.
Gram negative bacteria possess the ability through a series of Two Component Signal Transduction Systems (TCS) to remodel their own LPS cell wall constituents in response to stresses that impact the integrity of the cell envelope. This ability to sense/respond/modify and therefore stabilize their cell membranes and physiology assures their survival amidst harsh and varied pharmaceutical manufacturing environments (Nikaido, 2003; Raetz et al., 2007; Capra and Laub, 2012; Li et al., 2012; Bonnington and Kuehn, 2016; Sohlenkamp and Geiger, 2016; Bertani and Ruiz, 2018; Norris et al., 2018).
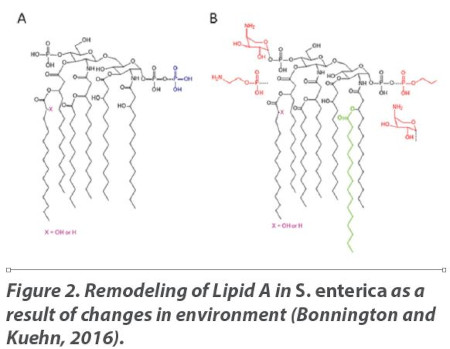
Figures 2A and 2B represent one such biochemical change in LPS fine structure by Salmonella enterica resulting from a shift in environmental conditions (Bonnington and Kuehn, 2016). Figure 2A depicts the Lipid A structure in an environment that contains 1mM Mg++ at pH 7.6. Lipid A is highly anionic, and the Mg++ in the external environment helps to neutralize the LPS net negative charge and stabilize the membrane (“salt bridges”). However, when the growth conditions shift to 10μM Mg++ and a pH of 5.8, the organism’s Lipid A loses the stabilizing effects of environmental magnesium and its TCS works to regulate the transcription of LPS-associated genes and generate a remodeled LPS which re-establishes membrane stability. In this case, the organism substitutes 4-amino-4-deoxy-L-arabinose (L-Ara4N) and zwitterionic phosphoethanolamine (pEtN) to stabilize the membrane (“sugar bridges”) and assure survival (Figure 2B).
The impact of various endotoxin fine structures on reaction with components of the native LAL cascade is relevant to the validation of these analytical methods. It follows that the reactivity of the recombinant reagents with likely contaminants must also be confirmed. Thus if we are to compare apples to apples, the only way to assure that patient safety decisions made using recombinant reagents are comparable to the compendial method is to perform the requisite studies on product samples containing quantifiable endotoxins autochthonous to their respective manufacturing operations.
Gram negative bacteria are ubiquitous and can be found in a very broad spectrum of environmental niches. It is possible that bacteria found in relatively nutrient rich environments such as a farmyard or mammalian gut, and those found in oligotrophic waters often possess significantly different LPS structural and biochemical characteristics. Similarly, endotoxins autochthonous to the pharmaceutical or medical device manufacturing environments will exhibit materially and analytically distinctive LPS fingerprints due to adaptations to their low nutrient and harsh environments (Morita, 1997). Sources of such endotoxins include (but are not limited to) any water that may purposefully or inadvertently access the manufacturing process from poorly designed or unvalidated water systems, lack of preventive maintenance or inadequate process controls (FDA, 1985). For example, in the 2015 FDA Inspection Guide section 7356.002A it states, “The quality of the water, and its endotoxin levels and controls, used in the upstream process should also be evaluated in order to ensure the removal of bacterial endotoxin to the appropriate level downstream.” While most pharmaceutical and medical device manufacturing sources of Gram negative bacteria often share a common oligotrophic nature and environmental condition (e.g. poor nutrients), such sources may vary, and each should be carefully assessed for its risk of product contamination.
Model Microorganism Approaches in Endotoxin Testing
Model organisms have been used in microbiological evaluation of specific types of processes for decades. An example of a model organism being used with significant success can be found in the development and validation of bacterial retention (sterilizing) filtration processes using Brevundimonas diminuta (specifically ATCC 19146) which can approach 0.2μm in its shorter dimension. This Gram negative organism has been used as a model because of its small size and, thus, it presents a reasonably rigorous challenge to a 0.2μm rated membrane filter. Obviously, this organism would be too large to use in the evaluation of viral retention filters and too small for use as a quality control test with pore size rated larger membranes. However, with the narrow pore size rating of filters, it is a useful model organism.
With respect to endotoxins testing, it is logical that the more common a Gram negative bacterium is as a contaminating microorganism in parenterally administered products, the better its endotoxin would serve as a “model” analyte. It has been known since the pioneering work of Florence Seibert and her colleagues as early as 1923 that the “filterable and pyrogenic” substance in water used to prepare intravenous therapies originated from a Pseudomonad isolated from the water systems (Seibert, 1923). However, the selection of E. coli as a model calibration analyte was an illogical choice for the pharmaceutical industry because finding of coliform bacteria within the healthcare product manufacturing industry is extremely rare. In fact, we know that there are a number of strains of non-fermenting bacteria that are often recovered from pharmaceutical manufacturing facilities, most notably pharmaceutical waters used to prepare Water for Injection (Reid, 2019) and we use the plural form of the term “endotoxins” because of the high level of variability in fine structures of LPS that can initiate both the innate mammalian response and the natural LAL cascade.
Therefore, the selection of a single Gram negative “model” endotoxin sourced from a single species or strain of bacteria used for tasks other than reagent calibration or product-specific suitability would be logical only if all Gram negative bacteria likely to contaminate parenteral products or implantable medical devices had identical endotoxin structures.
Because of this variability, it is unlikely that any laboratory can replicate the exact and often variable environmental conditions under which autochthonous microorganisms adapt and, in the process, remodel the structures of their lipopolysaccharides. RSE and CSE are laboratory constructs, meaning that they are prepared from selected strains of E. coli grown in the laboratory under well documented and well controlled conditions (Poole, 1997). Once grown, the LPS that results from those specific conditions is extracted using a hot phenol (Westphal) extraction method. The extracted LPS is then further formulated with lactose and polyethylene glycol and is freeze dried. While this well-controlled process may be suitable for large batches of a stable form of LPS meant for assay calibration purposes, it is biochemically dissimilar from the variety of microorganisms and naturally remodeled LPS structures that are found in pharmaceutical/medical device manufacturing environments. Therefore, the original intention to have RSE (CSE) be representative of all potential sources of the diversity of autochthonous endotoxins found in pharmaceutical operations was unrealistic and based upon current knowledge, clearly mistaken. It is therefore necessary to confirm that alternative endotoxin assays can detect such autochthonous endotoxins generated under what we could term “real world conditions”. In sum, we need a realistic understanding of an alternative method’s ability to detect a range of endotoxins not just a highly purified and concentrated endotoxin selected from a species unlikely to reside in our environments. It is necessary to consider what we know of endotoxin contamination events which have occurred in healthcare products.
Known Endotoxin Contamination Events
The pharmaceutical industry has worked diligently to reduce or eliminate endotoxins contamination. However, the contamination of drug products and medical devices with bacterial endotoxins continues to be reported with profound impact to patients and market supply. In the majority of cases endotoxins contamination has been associated with raw materials, APIs, manufacturing process water, sources of stagnant water and/or a breach of established manufacturing control. In all circumstances it is clear that endotoxins contamination compromising product quality originates from microorganisms autochthonous to the manufacturing facilities of the components, raw materials (excipients and drug substances, device components) and final products. Below are examples of reported events of a wide range of products contaminated with endotoxins autochthonous to their manufacturing facilities.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
FDA Observations and Warning Letters
Upon researching compliance issues from February 2011 through to March 2019, we found that FDA issued 13 warning letters and 66 observations to global pharmaceutical and medical device manufacturers regarding endotoxins excursions, testing and control.
Of these, 90% were associated with human drugs and biologics and approximately two thirds of those were attributed to the manufacturing facility. The majority of FDA inspectional observations associated with the manufacturing facility included reference to raw materials, manufacturing process controls, source water and water system defects. Five observations are especially noteworthy because they illustrate endotoxins contamination associated with autochthonous sources.
- An inspection of the firm CAF-DAF, of Neder-over-Heembeek, Belgium in May 2013 included reference to 44 batches of drug product rejected due to high endotoxins associated with a deficient cleaning program.
- In June 2013 an inspection of GlaxoSmithKline Biologicals, Dresden, Germany noted in the observations 41 occurrences of endotoxins limit excursions attributed to raw materials. However, there were also 16 occurrences of water ingress into the manufacturing buildings that were inadequately assessed by the firm as potential sources of contamination.
- Observations from the inspection of ID Biomedical, a subsidiary of GSK Biologicals, St. Foy, Canada in April 2014 referenced multiple product lots failing the endotoxin specification with activity levels exceeding 60,000 EU/mL. Both Stenotrophomonas maltophilia and Pseudomonas species were also frequently recovered, and the firm’s maintenance and monitoring of cleaning was identified as being inadequate.
- Endotoxins contamination of multiple drug product lots were attributed to contaminated drug substance as recorded in observations associated with an inspection of Cubist Pharmaceuticals, Lexington, USA in July 2014.
- In April 2015 inspectional observations for CenexiS, Fontenay Sous Bois, France included reference to endotoxins originating from raw materials or the inadequate cleaning of product storage vessels.
It must be noted that the above observations also frequently included the FDA’s opinion that the firm had failed to complete adequate investigations. Nevertheless, the content of these observations illustrates the commonality of endotoxins contamination originating from sources autochthonous to the manufacturing environment.
Peritoneal Dialysis Solutions
In December 2010 a number of patients receiving peritoneal dialysis solution manufactured at Baxter’s Castlebar facility experienced aseptic peritonitis (EMA, 2011). Initial investigation determined the cause to be endotoxins contamination originating from two manufacturing vessels. These two vessels had cracks serving as conduits to a hollow leg support that created a cavity containing a reservoir of stagnant fluid. Peritoneal dialysis formulations and process waters flowed bidirectionally between the structural cavity and the product and introduced endotoxins from the microorganisms autochthonous to that reservoir. Gram negative microorganisms namely Stenotrophomonas maltophilia and Sphingomonas paucimobilis were recovered from the source. Peritoneal dialysis formulations and the process waters associated with this event are not nutrient rich and result in endotoxins peculiar to those autochthonous sources. The European Medicines Agency’s scientific conclusions were ‘Overall, a combination of factors may have played a role in the findings, such as inadequately designed equipment, mechanical failure of equipment (cracks having been detected in some tanks used in the affected lines), methods of microbial monitoring not applied routinely and not state of the art, including outdated methods for monitoring endotoxins, and an inadequate cleaning and sanitization regimen’. This clearly underscores the relevance of endotoxins autochthonous to manufacturing facilities, their control and especially a robust and effective method of detection and quantitation.
Medical Devices
The testing for bacterial endotoxins in/on implantable medical devices is a critical requirement especially in cases where those products serve as replacements of heart valves or vascular conduits in patients of all ages. However, there was a disastrous example of a manufacturer that was negligent in their understanding of the consequences of bacterial endotoxins that were adulterating several of their products. It was eventually determined from clinical reports of patient fatalities and a series of FDA inspections that their facilities had significant GMP deficiencies in their manufacturing processes (FDA News, 2007). The FDA inspectional observations for this firm documented that many of their pediatric heart values and conduits were contaminated with bacterial endotoxins in the range of 51-84 EU/device along with sterility test failures which were ignored and that the product lots were subsequently released. This reported range of endotoxins contamination is significant because the USP requirement for an implantable device is less than 20 EU/device. A year after the FDA executed a court order for a mass seizure of $30 million of their product that included more than 20 different product types, The Journal of Thoracic and Cardiovascular Surgery (Carrel et al, 2008) subsequently reported that this firm’s products had a history of harmful patient effects, many resulting in death. It was reported in their published results “Retrospectively, the main findings were persistent fever or sub-febrility over months and a halo-like enhancement on computed tomographic scans, extensive microbiological examinations were performed without finding a causative organism” (Carrel et al, 2008). Febrile reactions originating from bacterial endotoxins associated with product contamination occur from endotoxin being present in product rather than microbial proliferation in product pre- or post- injection or installation. In this case the finding of a sterile lesion indicates the patient febrile responses arose from autochthonous endotoxin on the product arising from inadequate in process controls.
In the court records FDA filed against this firm and by personal account from one of the authors on this paper who was a coinvestigator and established expert witness during the court hearings, it was established that the firm’s procedures for testing the microbial quality of their water used to process products did not include analysis for the presence of bacterial endotoxins, even though its device specifications required that it not exceed the USP allowable endotoxin limit for an implantable device. Their process water was used to prepare various solutions that came in direct contact with raw materials, components, and finished devices. The risk from endotoxins derived from autochthonous sources during the manufacturing of their finished products was compounded by their lack of a process control to remove them in subsequent manufacturing steps. (US District Court, NJ, Civil Action, April 16, 2007).
Gentamicin
In 1998, CDC received multiple reports across numerous states describing febrile patient reactions associated with an off-label once daily dosing (bolus) regimen of two sources of gentamicin (CDC, 1998). Fanning et al. (2000) reported on the FDA investigation which revealed that 10 percent of gentamicin lots tested had elevated endotoxin levels (>1.7 EU/mg), and an additional 4 percent of the lots possessed levels above the acceptable threshold with once daily dosing. The two products implicated originated from the same supplier of bulk gentamicin. At the time, the FDA laboratory tested numerous products associated with the hospital bound patients with high fever symptoms.
It cannot be overstated, that the sensitivity and detectability of the bacterial endotoxin LAL method used on all products tested were critical for the traceback of the contaminated lots to the specific manufacturing source. The LAL gel-clot results were clearly indicative of the gentamicin source of the API made in China (Personal communication with the FDA analyst and co-author on this article). Inspection of the bulk supplier from the targeted manufacturer revealed numerous cGMP violations that likely resulted in material contaminated with endotoxin from Gram negative bacteria autochthonous to the manufacturing facility. Friedman (2005) listed the cGMP violations which included the use of inappropriate quality water for processing and cleaning activities.
Conclusion
Effective endotoxins tests are essential to the assurance of patient safety. There is irrefutable evidence that the compendial tests described in USP<85> have served well since they were introduced over four decades ago. However, both reported and unreported occurrences of endotoxins contamination of raw materials, medical device and pharmaceutical manufacturing operations continues. Out of specification endotoxins findings in product, although infrequent, illustrate the sources, process control breaches and malfunctions that may result in contamination that could impact patients.
These endotoxins sources have in common an environment that is generally oligotrophic and extremely nutrient poor when compared to the typical laboratory culture medium. Research further indicates that the Gram negative bacteria autochthonous to these oligotrophic environments possess lipopolysaccharides (LPS) that are chemically different from calibration standards purified from laboratory-grown coliforms and so called ‘naturally occurring endotoxins’ (NOE). We also know that these autochthonous bacteria may remodel their LPS in response to the nutrient poor environments to which they must adapt. We conclude that a sole reliance on highly purified LPS calibration standards derived from coliforms, while perhaps meeting the product-specific controls for method suitability, is insufficient to demonstrate comparability to existing <85> methods (Akers et al., 2020b). It then logically follows that the use of recombinant reagents, which are very different than naturally sourced Limulus amebocyte lysate (LAL), demands an antecedent demonstration of their comparability to detect and quantify endotoxins that realistically have the potential to contaminate product, that is, endotoxins autochthonous to a manufacturing facility.
Based on the data in the public domain and current research literature, we can only conclude that the proposed alternative recombinant reagent-based endotoxins tests, have not yet adequately demonstrated comparability to USP <85> for their ability to accurately detect and quantify autochthonous endotoxins.
Literature Cited
Akers, James, Dennis Guilfoyle, David Hussong, Karen McCullough, Robert Mello, Donald Singer, Edward Tidswell, Radhakrishna Tirumalai. 2020a. Functional Challenges for Alternative Bacterial Endotoxins Tests. Part 1. Attributes for Alternative Tests. Am. Pharm. Rev. 23 (4), 76-81.
Akers, James, Dennis E. Guilfoyle, David Hussong, Karen McCullough, Robert Mello, Donald Singer, Edward Tidswell, Radhakrishna Tirumalai. 2020b. Functional Challenges for Alternative Bacterial Endotoxins Tests. Part 2. Comparability. Am. Pharm. Rev. 23 (5), 18-27.
Bertani B, Ruiz N. Function and Biogenesis of Lipopolysaccharides. EcoSal Plus. 2018;8(1): doi:10.1128/ecosalplus.ESP-0001-2018
Bonnington, Katherine E. and Meta J. Kuehn. 2016. Outer Membrane Vesicle Production Facilitates LPS Remodeling and Outer Membrane Maintenance in Salmonella during Environmental Transitions. mBio. 7(5): 1-15. doi:10.1128/mBio.01532-16.
Capra, Emily J and Michael T. Laub. 2012. Evolution of Two-Component Signal Transduction Systems. Annu. Rev. Microbiol. 2012; 66: 325–347. doi:10.1146/annurev-micro-092611-150039
Carrel, T.P., F. S. Schoenhoff, J. Schmiidli, M. Stalder, F. S. Eckstein, and L. Englberger. “Deleterious Outcome of No-React-treated stentless valved conduits after aortic root replacement: Why were Warnings ignored?”. The Journal of Thoracic and Cardiovascular Surgery, 2008; 136: 52-57
Centers for Disease Control and Prevention (CDC). Endotoxin-like reactions associated with intravenous gentamicin--California, 1998. MMWR Morb Mortal Wkly Rep. 1998;47(41):877-880
European Medicines Agency. 2011. Assessment report for Dialysis solutions produced at Castlebar by Baxter group of companies and associated companies Procedure number: EMEA/H/A-31/1290
https://www.ema.europa.eu/en/medicines/human/referrals/baxterdialysis-solutions
Fanning MM, Wassel R, Piazza-Hepp T. Pyrogenic reactions to gentamicin therapy. N Engl J Med. 2000;343(22):1658-1659. doi:10.1056/NEJM200011303432218
FDA News, P07-69, April 17, 2007, “FDA Seizes All Medical Products from N.J. Device Manufacturer for Significant Manufacturing violations”,
https://www.hpra.ie/docs/default-source/Safety-Notices/sn2007(01)_shelhighimplantabledevices_v1825_attach01_100507.pdf? sfvrsn=0
Food and Drug Administration. 1985. Inspectional Technical Guidance, No. 40 “Bacterial Endotoxins/Pyrogens. https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens
Food and Drug Administration. 2015. Program 7356.002A. CHAPTER 56 – DRUG QUALITY ASSURANCE “Sterile Drug Process Inspections.” Section 3.7(1), page 19 https://www.fda.gov/media/75174/download
Friedman, R.L. Aseptic Processing Contamination Case Studies and the Pharmaceutical Quality System. PDA Journal of Pharmaceutical Science and Technology Mar 2005, 59 (2) 118-126.
Li, Yanyan, Daniel A. Powell, Scott A. Shaffer, David A. Rasko, Mark R. Pelletier, John D., Leszyk, Alison J. Scott, Ali Masoudi, David R. Goodlett, Xiaoyuan Wang, Christian R. H. Raetz, Robert K. Ernst. 2012. LPS remodeling is an Evolved Survival Strategy for Bacteria. Proc. Natl. Acad. Sci. 109(22): 8716-21.
Morita, R.Y, 1997, “Chapter 7 Physiology of Starvation-Survival”, In book: Bacteria in Oligotrophic Environments, Starvation-Survival Lifestyle, Chapman & Hall Microbiology Series, page 323
Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67(4): 593–656. http://dx.doi.org/ 10.1128/MMBR.67.4.593-656.2003.
Norris MH, Somprasong N, Schweizer HP, Tuanyok A. 2018. Lipid A remodeling is a pathoadaptive mechanism that impacts lipopolysaccharide recognition and intracellular survival of Burkholderia pseudomallei. Infect Immun 86:e00360-18.
https://doi.org/10.1128/IAI.00360-18.
Poole, S., P. Dawson, R.E. Gaines Das. 1997. Second international standard for endotoxin: calibration in an international collaborative study. J. Endotox Res. 4(3): 221-231
Raetz CR, Reynolds CM, Trent MS, Bishop RE. 2007. Lipid A modification systems in Gram-negative bacteria. Annu. Rev. Biochem. 2007. 76:295–329. http://dx.doi.org/10.1146/annurev.biochem.76.010307.145803.
Reid, Nicola. 2019. A global perspective for quantifying all endotoxins within pharmaceutical water systems. Presented at PharmaLab, Dusseldorf, Germany.
Seibert, F.B. 1923. Fever producing substances found in some distilled waters. Am. J. Physiol. 67: 90-104.
Sohlenkamp, Christian and Otto Geiger. 2016. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiology Reviews. 40(1): 133-159. https://doi.org/10.1093/femsre/fuv008
Trent, Stephen M., Christopher M. Stead, An Tran and Jessica V. Hankins. 2006. Diversity of endotoxin and its impact on pathogenesis. J. Endotox. Res. 12(4): 205-223.
US District Court, NJ District, Civil Action 07-CV-01769 Verified Complaint for Forfeiture IN REM, 2007, page 7 of 15, https://www.fda.gov/media/70211/download