Cell Therapy Development & Operations
Cell Therapy Development & Operations
Research & Early Development
Abstract
The generation of Chimeric Antigen Receptor (CAR)- and engineered T Cell Receptor (TCR)-expressing cell therapies from patient lymphocytes commonly relies on viral vectors for transgene delivery. A number of processing steps aid in size reduction and clearance of residual DNA species, however, despite these process-level controls, nucleic acid impurities in the vector product may be carried over into the CAR-T drug product manufacturing process. Residual DNA impurities are a critical quality attribute (CQA) due to the potential risk for infectivity and immunogenicity. Although the development of biologics and vaccines have laid some of the analytical groundwork for their detection and quantification, distinguishing between exogenously introduced DNA species and human DNA from the cell therapy drug product is a key challenge. Two newer exemplary analytical approaches - TapeStation™-based sizing and next generation sequencing - are described, along with a discussion of how outputs from these complementary approaches can inform assessments of residual DNA impurity risks in cell therapies.
Introduction
Chimeric Antigen Receptor (CAR) and Engineered T cell Receptor (TCR) Technology
T cells play important roles in adaptive immunity by serving as sentinels against foreign pathogens. T cells that have been engineered to recognize cancer-specific antigens present a promising treatment option for a number of hematological cancers and solid tumors.1,2
Potential cellular modifications include vector transduction with Chimeric Antigen Receptor (CAR) or engineered T Cell Receptor (TCR) transgenes to boost binding affinities to tumor antigens and trigger activation, proliferation, and targeted killing of cancer cells.
In recent years, both CAR and TCR immunotherapies have produced encouraging clinical outcomes.3–5
Gene Editing Materials Used in Cell Therapy Manufacturing May Contribute to Residual DNA Species in the Cell Therapy Drug Product
Despite different mechanisms of immunorecognition, both autologous CAR- and TCR-based therapies require ex vivo editing of a patient’s own T cells. The cell therapy drug product manufacturing process is complex and involves a number of critical raw materials, each with their own manufacturing processes. For example, the CAR or TCR transgene is commonly delivered via viral vector. During transduction, the viral vector product is a major source of DNA impurities. As a critical component, the vector product includes upstream (cell culture, plasmid transfection) and downstream (vector purification) processes.6 In upstream vector production, a HEK293-derived producer cell line is transfected by three or four plasmids carrying viral structure and replication genes along with the desired CAR or TCR transgene. Expression of these genes results in vector assembly. Viral production cells are lysed and the assembled vector particles are subsequently harvested.7,8 Within the downstream vector process, the desired vector product is isolated and impurities such as DNA from the plasmids and producer cells are generally reduced in size and quantity by process steps that may incorporate nuclease treatment, chromatography, and filtration and buffer exchanges.7–9 After the vector is added to the cell therapy drug product process (during the gene transfer step), washes and media exchanges in subsequent steps like expansion or post-transduction selection contribute to further reduction in the levels of DNA impurities. Despite the clearance, some level of residual DNA may be carried through to the final drug product. Residual DNA impurities are a critical quality attribute (CQA) due to their potential impact on product quality and safety. Analytical methods to characterize these impurities are critical to enhance process and product understanding.
Risks of Residual DNA Impurities
Residual DNA impurities may pose safety concerns for therapeutics and vaccines. Risks include oncogenicity,5,10 infectivity,11 and immunogenicity.6,12 These risks exist for both vectors that integrate within the host cell genome (such as lentivectors) and nonintegrative vectors, such as adeno-associated virus (AAV) vectors. For integrative vectors, the potential for oncogenicity arises through insertion of exogenous DNA sequences near proto-oncogenes or tumor suppressor genes or through the expression of oncogenic sequences.11 In the case of AAV, indiscriminate packaging of extraneous DNA sequences other than the intended vector genomes may occur.13 Encapsidated DNA impurities are theoretically delivered directly into the target cell along with the intended transgene payload and thus could have an increased chance of integration or expression due to spatial proximity to genomic DNA. Depending on the lineage of the producer cell line, viral sequences such as those encoding for adenovirus E1A and SV40 large T antigen may be present as DNA impurities and pose an infectivity risk.11 Lastly, immunogenicity may arise due to induction of innate immunity by the DNA fragments themselves (especially in complex with proteins) or due to the expression of exogenous viral or plasmid DNA, whose products may be recognized by Toll-like receptors.12
Regulatory perspectives on residual DNA impurities are captured in two key publications: the FDA’s 2020 Guidance for Industry14 and the WHO’s 1998 Expert Committee on Biological Standardization.15 Within these guidance documents, the proposed limit is 10 ng of residual cellular DNA per dose of injected drug product with DNA fragmentation to achieve an average size of 200 base pairs.
Risk Mitigation Strategies
To reduce safety risks posed by DNA impurities, the amounts and sizes of residual DNA impurities should be limited and controlled. Within the context of cell therapy manufacturing, process control of DNA impurities includes minimizing the introduction of vector-derived DNA impurities into the cell therapy manufacturing process at the outset (via vector product release specifications), and reduction of impurities via downstream processing steps. After vector transduction of T cells, washes and media addition and exchange steps (such as those used during T cell expansion and post-transduction cell selection) may contribute to a sizable reduction in the quantities of DNA impurities. Quantifying the clearance of vector-derived DNA in the drug product process poses a major analytical challenge: the T cell-based drug product contains human DNA and a high degree of overlap is expected between patient-derived DNA and exogenous DNA impurities from the vector producer cells which are also human in origin. This challenge highlights the importance of leveraging appropriate analytical technologies that aid in the characterization of these impurities in the vector product.
Analytical Methods for Characterizing Residual DNA Impurities
Aside from process controls, characterization of the size distribution and identity of residual DNA species can inform the assessment of impacts to product quality and safety. The abundance and clearance of exogenous DNA impurities can be evaluated using a number of well-established technologies.16 Among the available technologies, molecular methods such as qPCR or ddPCR are the most commonly used and offer the best balance between throughput and sensitivity.17 Both approaches use PCR amplification and detection of a target amplicon within either the plasmid or host cell genome as a surrogate for residual plasmid or host cell DNA levels in the sample. For residual host cell DNA assays, the selected amplification target is often derived from a multicopy gene to increase assay sensitivity.18 Commercially available kits, designed with targets that are optimized for a number of host species, are available for the detection of residual host cell DNA. These types of qPCR- and ddPCR-based assays are commonly used for in-process testing and as product release assays.
One drawback of PCR-based methods is that the quantification of DNA impurities relies on the detection of intact target amplicons. As mentioned, the quantification of the target amplicon is used as a surrogate for the levels of total host cell DNA or plasmid DNA impurities. Although this may be an accurate assumption for DNA that is relatively intact, undercounting may occur for DNA that has been fragmented by nuclease treatment or otherwise degraded. A second analytical limitation is that the detection of a single target amplicon does not provide precise sizing information. Theoretically, all species that contain the target amplicon will be detected however the quantification associated with a single target only provides a lower sizing bound. Reporting PCR-based quantification readouts from multiple, nested amplicons can provide some sizing information but it is not possible to obtain a continuous size distribution profile of DNA impurities using PCR-based techniques. In the cell therapy vector product, larger DNA fragments are suggested to pose higher oncogenicity and infectivity risks19 so assessing process clearance of larger DNA fragments and demonstrating reduction of average DNA size through the process is a key objective to mitigate risk. These shortfalls of PCR-based approaches for assessing DNA impurities spotlight the need for additional tools for understanding the risks posed by these impurities in the vector product.
Analysis of Size Distributions of DNA Impurities in Vector Preparations
The TapeStation™: a gel-based separation technology
The TapeStation platform (Agilent Technologies) is a miniaturized automated electrophoresis system that separates DNA fragments by size and can also be used to determine their relative abundances. The system consists of a self-contained consumable, called a ScreenTape, that holds a sieving matrix. A proprietary sample buffer contains a dsDNA-sensitive fluorescent dye (SYBR™ Green I) and two reference markers. The buffer is mixed with the samples of interest as well as a DNA ladder sample prior to separation and detection. Several configurations of the ScreenTape are available depending on the sizing range of interest. An application note from Agilent reports the utility of the system in assessing genomic DNA removal in biologics process development using the genomic DNA ScreenTape.20 In addition to the genomic DNA TapeStation assay, which separates DNA species between 200 and 60,000 base pairs in length, several additional configurations are available for examining smaller DNA impurities. For cell therapy applications, the TapeStation D1000 and D5000 assays may be suitable for assessing the size distribution of DNA impurities in the vector product. Depending on the assay kit, impurities ranging from 35 to 5000 bp may be analyzed.21,22 There are high sensitivity versions of both the D1000 and D5000 products (denoted D1000HS and D5000HS) which offer improved sensitivity (as low as 5 pg/μL for a single peak) albeit at the cost of lower assay robustness since the HS assays have lower tolerance for contaminating salts and are more prone to matrix interference.
Assessment of the TapeStation for the Characterization of DNA Impurities
Application of vector samples to the TapeStation platform requires a column-based DNA extraction with a DNA or PCR purification kit to reduce matrix interference and to isolate DNA. Extracted samples are then mixed directly with the assay reaction buff er. Analogous to a traditional agarose gel, a separate DNA ladder sample is also prepared by mixing with the reaction buffer. The ladder sample is run in a separate lane, alongside each set of samples to provide comparative sizing. The two reference markers in the reaction buffer (one upper marker and one lower marker) flank the sizing range and enable proper alignment of the sample and ladder electropherograms. In each sample trace, the peak area of the upper reference marker is also used to normalize the fluorescence signal and to assign concentrations to the resulting electropherogram peaks.
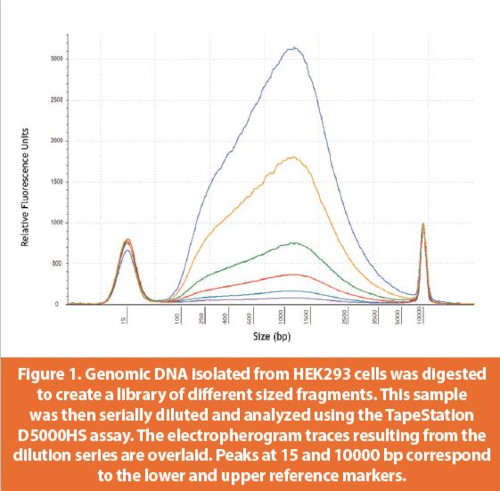
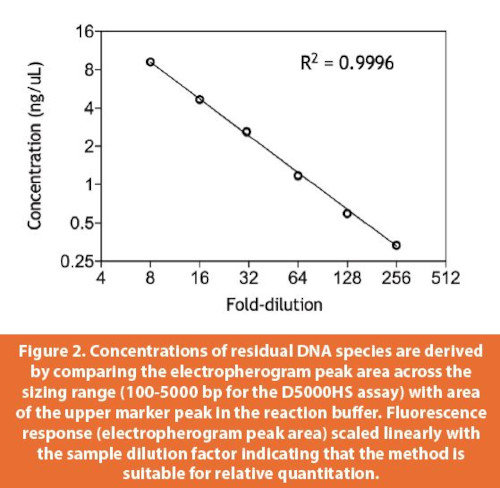
The TapeStation assay sensitivity was determined based on dilution series of dsDNA fragments of defi ned lengths (300 and 1500 bp for the D1000 and D5000 high sensitivity assays, respectively).21,22 Per Agilent’s D5000 Technical Note, signal to noise ratios greater than 3 were reported for single dsDNA species as low as 5 pg/μL. When the DNA sample consists of a mixture of species, the limit of detection rises since the electropherogram transforms from a single, discrete peak into a smear and baseline resolution of peaks is lost. To better determine assay performance with heterogenous mixtures of DNA fragments, a library was generated by subjecting HEK genomic DNA to partial nuclease digestion. This library of fragments was serially diluted prior to analysis in the D5000HS assay (Figure 1). Comparison of the electropherogram peak areas to an internal standard (upper reference marker) peak area produced a DNA concentration in ng/μL for each dilution. These concentrations were then plotted across the full dilution range (Figure 2). Excellent linearity was observed suggesting that relative quantitative comparisons across samples is possible. The lower limit of detection for the heterogenous DNA sample was ~300 pg/μL.
Rigorous assessment of quantitative accuracy is challenging with a dilute, heterogenous mixture. Here, accuracy was roughly gauged by comparing concentration values from the TapeStation with another dye-based method (Qubit dsDNA HS Assay Kit). The TapeStation D5000HS and Qubit concentration values were generally within 30% however, for a few samples, the values deviated by as much as 50%. Further assessment of DNA abundance, perhaps by using an orthogonal third method, may provide a more complete comparison of the quantitative accuracy of different approaches.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
Sizing accuracy was determined using a 200 bp dsDNA standard and the ladder sample. The D5000HS specification of sizing accuracy +/- <15% was confirmed in our studies with both the 200 bp dsDNA standard and differently sized species in the ladder sample (ranging from 100 to 5000 bp). For a mixture of DNA fragments (smear in the electropherogram), the TapeStation software reports an average size weighted by the electropherogram peak area. Decreasing size distributions due to increased nuclease activity are captured in TapeStation electropherograms, as shown in Figure 3. The ability of successive vector process steps to reduce the average size and relative abundance of the residual impurities may provide justification of process suitability and robustness and demonstrate sufficient mitigation of DNA impurity risks.
Overall, several features of the TapeStation make it a valuable tool for characterizing residual DNA impurities associated with vector samples. The sensitivity of the assay is compatible with residual DNA levels in the vector product. The internal reference markers, used for electropherogram alignment and signal normalization, enable assessment of the average size of DNA impurities and comparisons of the size distributions between samples. Lastly, the assay throughput of up to 96 samples per run makes it well suited to support vector process development. Demonstrating reduction in average DNA size is an important risk mitigation strategy for DNA impurities. The application of TapeStation-based analytics both in-process and at the final vector product stage can help inform the assessment of vector-associated residual DNA risks.
Sequencing-Based Assessment of DNA Impurity Risks
As previously discussed, larger DNA impurities are thought to carry higher risks of oncogenicity and infectivity.19 The severity of risks posed by DNA impurities may also be stratified based on sequence. For instance, the majority of host cell DNA impurities from HEK-derived cell lines are human in origin and their expression is unlikely to cause immunogenicity. On the other hand, infectivity and immunogenicity risks may arise due to the presence and expression of viral and bacterial DNA sequences found within the plasmids or producer cell genome.11 To control these risks, assays that monitor levels of particular viral genes or proteins may be employed.

Another approach for characterization is to use high-throughput sequencing to define the composition of DNA impurities and assess the degree of enrichment of particularly problematic sequences. This can be achieved by comparing read alignments and read counts from a vector-derived DNA sample with DNA extracted from a non-transfected control. A normalized mapping frequency provides the number of mapped reads per length of a reference sequence. Elevated copy numbers of a particular reference sequence are reflected in elevated normalized mapping frequencies. For example, within a vector-derived DNA sample, normalized mapping frequencies for plasmid are expected to be higher relative to nuclear chromosomes due to higher copy numbers of plasmid added during transfection. The normalized mapping frequency provides a means for comparing relative enrichment. Evaluation of normalized mapping frequency between vector-derived DNA and non-transfected controls addresses whether sequence enrichment is occurring during transfection and throughout the vector production process. A lack of enrichment of sequences with higher oncogenicity and infectivity potential (such as Ad5 and SV40 viral genomes) in the transfected sample can provide confidence that the manufacturing process is not contributing to an elevation of the oncogenicity and infectivity risks of residual DNA impurities in the vector product. In conjunction with models developed to assess oncogenic and infectivity activity of DNA,11 relative lengths and abundances of high-risk species can be used to determine a safety factor with respect to an infectious or oncogenic event and inform overall DNA impurity risks in the product.
Summary
Residual DNA fragments are process-related impurities that present several potential risks including oncogenicity, infectivity, and immunogenicity. Within cell therapies, measurement and characterization of residual DNA species pose unique analytical challenges due to interference from patient-derived DNA in the drug product. Analysis of DNA impurities in the vector product provides a mechanism for upstream control, characterization, and risk mitigation. A number of well-established tools for DNA analysis (including molecular approaches like ddPCR and qPCR) are routinely employed in the vaccine and biologics industries; however, the aim of this review is to highlight two exemplary newer approaches that may be suitable for DNA analysis within the cell therapy vector product. The first is the TapeStation platform for understanding the size and amounts of vector process-derived DNA impurities. The second is the use of next generation sequencing to detect high-risk sequences within the pool of DNA impurities and assess their enrichment. Together, these complementary tools can aid in vector-level characterization and control of DNA impurity risks, inform both vector and drug product process development, and ensure that an appropriate level of impurity clearance is achieved in the final drug product.
Author Biographies
Jennifer Hu is a Scientist at Bristol Myers Squibb where she specializes in analytical strategy and method development for cell therapies and vector products. Before joining BMS, she worked on various facets of process and analytical development of protein-based therapeutics at Genentech. Jennifer holds a PhD in Biological Chemistry from MIT.
Hai Yue is currently an Associate Director in Cell Therapy Development & Operations (CTDO) at Bristol Myers Squibb. Prior to joining BMS, he worked at Amgen Inc. as a Scientist in Process Development for four years. Dr. Yue holds a PhD in Biochemistry from Washington University in St. Louis. Timothy G. Johnstone is a computational biologist at Bristol Myers Squibb with over a decade of experience in the bioinformatics field. In addition to leveraging his background in genomics, RNA biology, immunotherapy, and gene regulation to derive biological insights from complex datasets,
Dr. Johnstone specializes in architecting workflows and systems which enable actionable, reproducible analysis of data from novel NGS assays.
Christopher Clouser is a Principal Scientist with Bristol Myers Squibb and has a background in molecular biology with a focus in Next Generation Sequencing applications development. Prior to working for Bristol Myers Squibb, he worked for Life Technologies performing methods and technology development to help launch the SOLiD and Ion Torrent sequencing platforms. In his current role, he leads a team focused on development of genomic methods to help understand CAR-T biology.
Taylor Zhang is a Director in Product and Analytical Department in Cell Therapy Development and Operations at Bristol Myers Squibb, in Seattle, Washington. Prior to Bristol Myers Squibb, he was a Principal Scientist and group leader in the Protein Analytical Chemistry Department at Genentech. He received his PhD in Analytical Chemistry from Iowa State University.
References
- Li D, Li X, Zhou W-L, et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct Target Ther. 2019;4(1):35. doi:10.1038/s41392-019-0070-9
- Guedan S, Calderon H, Posey AD, Maus M V. Engineering and Design of Chimeric Antigen Receptors. Mol Ther - Methods Clin Dev. 2019;12:145-156. doi:10.1016/j. omtm.2018.12.009
- Barrett DM, Grupp SA, June CH. Chimeric Antigen Receptor– and TCR-Modified T Cells Enter Main Street and Wall Street. J Immunol. 2015;195(3):755-761. doi:10.4049/jimmunol.1500751
- Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019;10. doi:10.3389/fimmu.2019.02250
- Russell WC, Graham FL, Smiley J, Nairn R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J Gen Virol. 1977;36(1):59-72. doi:10.1099/0022-1317-36-1-59
- Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017;31(4):317-334. doi:10.1007/s40259-017-0234-5
- Martínez-Molina E, Chocarro-Wrona C, Martínez-Moreno D, Marchal JA, Boulaiz H. Large-Scale Production of Lentiviral Vectors: Current Perspectives and Challenges. Pharmaceutics. 2020;12(11):1051. doi:10.3390/pharmaceutics12111051
- El Andari J, Grimm D. Production, Processing, and Characterization of synthetic AAV Gene Therapy Vectors. Biotechnol J. October 2020:2000025. doi:10.1002/biot.202000025
- Olgun HB, Tasyurek HM, Sanlioglu AD, Sanlioglu S. High-Grade Purification of Third-Generation HIV-Based Lentiviral Vectors by Anion Exchange Chromatography for Experimental Gene and Stem Cell Therapy Applications. In: ; 2018:347-365. doi:10.1007/7651_2018_154
- Sheng L, Cai F, Zhu Y, et al. Oncogenicity of DNA in vivo: Tumor induction with expression plasmids for activated H-ras and c-myc. Biologicals. 2008;36(3):184-197. doi:10.1016/j. biologicals.2007.11.003
- Sheng-Fowler L, Lewis AM, Peden K. Issues associated with residual cell-substrate DNA in viral vaccines. Biologicals. 2009;37(3):190-195. doi:10.1016/j.biologicals.2009.02.015
- Singh SK. Impact of Product-Related Factors on Immunogenicity of Biotherapeutics. J Pharm Sci. 2011;100(2):354-387. doi:10.1002/jps.22276
- Wright J. Product-Related Impurities in Clinical-Grade Recombinant AAV Vectors: Characterization and Risk Assessment. Biomedicines. 2014;2(1):80-97. doi:10.3390/biomedicines2010080
- US Food and Drug Administration. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs).; 2020.
- World Health Organization. WHO Expert Committee on Biological Standardization: Forty-Seventh Report.; 1998.
- Wang X, Morgan DM, Wang G, Mozier NM. Residual DNA analysis in biologics development: Review of measurement and quantitation technologies and future directions. Biotechnol Bioeng. 2012;109(2):307-317. doi:10.1002/bit.23343
- Wang Y, Cooper R, Kiladjian A, Bergelson S, Feschenko M. A Digestion-free Method for Quantification of Residual Host Cell DNA in rAAV Gene Therapy Products. Mol Ther - Methods Clin Dev. 2019;13(1):526-531. doi:10.1016/j.omtm.2019.05.005
- Himmelspach M, Gruber F, Antoine G, Falkner FG, Dorner F, Hämmerle T. Specific Quantitation of Genomic DNA in the Femtogram Range by Amplification of Repetitive Sequences. Anal Biochem. 1996;242(2):240-247. doi:10.1006/abio.1996.0459
- Yang H. Establishing Acceptable Limits of Residual DNA. PDA J Pharm Sci Technol. 2013;67(2):155-163. doi:10.5731/pdajpst.2013.00910
- Agilent Technologies. Agilent Solutions for Removal and Monitoring of Genomic DNA from Monoclonal Antibody Preparations.; 2013.
- Agilent Technologies. Performance of the Agilent D5000 and High Sensitivity D5000 ScreenTape Assays for the Agilent 4200 TapeStation System.; 2015. https://www.agilent.com/cs/library/technicaloverviews/public/5991-6356EN.pdf.
- Agilent Technologies. Performance of the Agilent D1000 and the Agilent High Sensitivity D1000 ScreenTape Assay for the Agilent 4200 TapeStation System.; 2016.