Last December, Vertex Pharmaceuticals and CRISPR Therapeutics shared initial clinical trial data from studies on CTX001, a cell-based therapy for sickle cell disease and beta-thalassemia. These diseases are characterized by defective or deficient production of hemoglobin meaning that patients require regular blood transfusions and cure is only possible through a high-risk bone-marrow transplant. CTX001 uses a clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) technology to knockout a gene resulting in re-establishment of levels of functional hemoglobin. The first two patients to have received this CRISPR-based therapy have remained blood transfusion independent a year after the initial treatment.1 This is just one example of the many clinical trials using CRISPR-based technologies that could eventually provide a treatment or cure for currently incurable diseases.
Despite these successes and the immense promise that CRISPR-Cas gene editing approaches hold, like all therapeutics, gene editing using CRISPR-Cas must prove to be safe and efficacious to be licensed. Concerns remain over the potential introduction of deleterious genome alterations by nuclease-based gene editors such as CRISPR- CRISPR associated protein 9 (Cas9), leading many to consider using base editors as an alternative approach to modify genes implicated in human disease.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!
CRISPR-Cas-Mediated Gene Editing
CRISPR-based technologies initiate DNA editing by generating DNA double-strand breaks (DSBs) at a specific region in the genome dictated by a guide RNA. In mammalian cells, DSBs are typically repaired in an error-prone fashion through the endogenous non-homologous end joining (NHEJ) repair pathway that leads to imprecise insertion and deletion (indels) of DNA base pairs at the repair site. Indels are useful in generating nonsense mutations leading to the loss of gene transcription and facilitating functional gene knockout. However, this approach is not precise enough to enable the introduction of specific changes within the DNA. Generation of a specific change at the target site requires the homology directed repair (HDR) pathway, which can be initiated by providing an exogenous DNA repair template delivered at the same time as generation of the DNA DSB by CRISPR-Cas (Figure 1). However, the HDR pathway is in constant competition with the NHEJ repair pathway, is inefficient, and is only effective in dividing cells. This prevents efficient application in post-mitotic cells which are often targeted for gene therapy. When working with patient-derived material to treat diseases such as sickle cell disease, a high number of the cells need to be edited precisely, which is not currently possible with HDR-mediated CRISPR to introduce specific genetic changes. The emergence of base editing, which does not rely on HDR or the generation of DNA DSBs to introduce single-nucleotide alterations may be set to revolutionize the field of cell and gene therapy.
Back to Bases
In 2016, two independent scientific groups developed base editing,2,3 a new and potentially transformative technology with the wherewithal to overcome many of the fundamental limitations of the traditional CRISPR-Cas9 system. Base editing employs deaminase enzymes to make a specific base-pair change in the DNA. The deaminase is recruited to the target site by a catalytically impaired form of Cas9 and a guide RNA and catalyzes the precise change in the DNA without the requirement of potentially harmful DSBs or inefficient HDR-mediated mechanisms. Two types of base editors have been developed, Cytidine Base Editors (CBEs), which mediate the conversion of C-G base pairs to T-A,2,3 and Adenine Base Editors (ABEs), which induce A-T to G-C conversion,4 allowing (theoretically) for the correction of all transition mutations (Figure 1). Since its introduction in 2016, base editing platforms have been developed and optimized to increase the efficiency, targeting scope and precision of the technology and this has expanded its potential applications both in the laboratory and the clinic.

Base Editing in Gene Therapy
Over 300 million patients are affected by rare diseases worldwide and many have no or limited treatment options available. Most of these are monogenic diseases caused by Single Nucleotide Variants (SNVs). Base editing, with its potential to correct SNVs, could provide efficient and safe one-time treatment for many rare diseases. It is estimated that base editors could correct about 37% of pathogenic SNVs, responsible for the development of 4000 diseases.5 Numerous in vitroand animal model studies provide proof-of-concept data with CBEs and ABEs for different gene therapy applications including phenylketonuria, Niemann-Pick disease (a form of neurodegenerative ataxia), hereditary deafness, Duchenne muscular dystrophy, cystic fibrosis and progeria (Table 1).
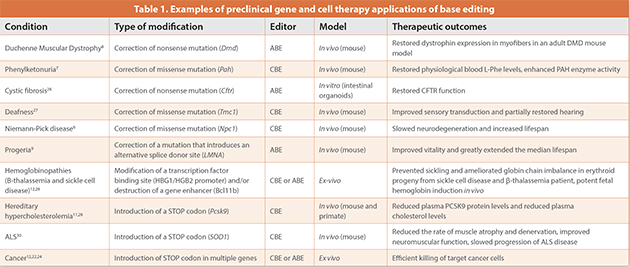
6–9 Progeria is a lethal and incurable disease of premature aging and patients with this disease typically survive only until their mid-teens. A group led by David Liu at the Broad Institute of Harvard and MIT used an ABE delivered as adeno-associated virus to treat a mouse model of progeria. They achieved the correction of the point mutation in the Lamin A gene, which is associated with progeria, and extended the lifespan of these mice almost three times compared with untreated controls.9
Base editors have also been used to alter mitochondrial DNA (mtDNA) in human cells in culture. Mitochondria do not have an RNA transport mechanism thereby prohibiting the use of a CRISPR-Cas base editor; Joseph Mougous and colleagues innovatively used Transcription- Activator-Like Effector Nucleases (TALENs) protein-based approach to target the deaminase to the mtDNA.10 The base editing of mtDNA is achieved without cleavage of the mtDNA as this would lead to its degradation. Such findings indicate that base editing could be used to treat Leber’s hereditary optic neuropathy, one of the most common mtDNA diseases characterized by the loss of retinal cells and sudden blindness.
Another example of the use of base editing as a gene therapy comes from the field of complex diseases. Naturally occurring inactivating mutations in the PCSK9 and ANGPL3 genes are associated with low levels of circulating cholesterol and triglycerides levels, respectively. These findings paved the way for the exploration of therapeutic strategies to achieve the same outcome for patients with familial hypercholesterolemia, who are at substantially increased risk of developing life-limiting cardiovascular disease. Verve Therapeutics announced in a press release11 that they had used an ABE delivered using lipid nanoparticles to edit and inactivate PCSK9 or ANGPL3 in liver cells in non-human primates. Circulating levels of lipids were durably reduced in the treated non-human primates, providing preliminary evidence that base editing could offer a safe“one-and-done” treatment for patients with this disease. Verve Therapeutics stated that they are seeking a first-in-human trial of this approach in 2022.
Although these data are exciting, gene therapies still face substantial challenges in terms of effective delivery to a variety of different tissues. One route employed to circumvent this limitation has been to modify genes in cells harvested from the patient and then transfuse the patient with their modified cells ushering in the era of cell-based therapeutics.
Base Editing in Cell-Based Therapies
As discussed at the start of this article, CRISPR-Cas gene editing approaches are being used to potentially cure patients with either sickle cell disease or β-thalassemia using a cell therapy approach. Care is needed with selection of the guide RNA used to disrupt the target gene, in this case BCL11A, to ensure unwanted DNA alterations are not introduced into the patient’s CD34+ hematopoietic stem and progenitor cells owing to Cas9-mediated DNA DSBs elsewhere in the genome. Base editors have also been explored to treat the same diseases. An ABE was used to introduce single base changes in the promoter sequence of HBG1 and HBG2 genes that encode fetal hemoglobin.12 These alterations introduced in CD34+ cells resulting in persistent expression of fetal hemoglobin in 60% of the edited cells, indicating that a base editor could be used to edit hematopoietic stem and progenitor cells in patients with these blood disorders with the advantage that DNA DSBs are not being introduced into the edited cells.
In 2017, the US Food and Drug Administration approved KYMRIAH®13 and YESCARTA®14 two CD19-targeted Chimeric Antigen Receptor (CAR)-T cell therapies, for the treatment of hematological malignancies, marking a milestone in the treatment of patients with cancer. Two additional CAR-T cell therapies, BREYANZI®15 and TECARTUS™,16 were approved more recently, yet despite these successes, the future of CAR-T cell therapy is still largely unclear. Will CAR-T cells therapies be an effective last line approach for a narrow range of indications, such as blood cancers and for a small number of patients or will they become more widely applied as frontline therapies, including for the treatment of solid tumors? Currently, ineffectiveness against solid cancers and the autologous nature of approved CAR-T cell therapies are the two biggest limitations to widespread application. Autologous CAR-T cell therapies are produced using a patient’s own T cells, which are genetically modified ex vivo to express a CAR (the molecule that recognizes cancer cells and instructs T cells to attack the tumor) before being infused back into the patient. The use of autologous cells has the advantage of reduced or absent immunological reactions to the therapy, but the manufacture of these cells has multiple limitations. The time required for manufacturing (at least three weeks) is often too long for critically ill patients and the quality of a cancer patient’s T cells might be compromised by the disease and by previous treatments, thereby jeopardizing effective manufacture of this personalized therapy. In addition, costs per patient are incredibly high.
The use of allogeneic CAR-T cell therapies, where T cells from healthy donors are engineered to create a bank of cells available on an ad hoc basis for multiple patients, could overcome these limitations and help to make immune cell therapy more widely available to patients with cancer or other diseases amenable to immune cell-based therapies. However, allogeneic CAR-T therapies require a higher level of genetic manipulation to become a safe and effective medicine. The immunological interactions between the host and the donor are responsible for potentially fatal Graft-Versus-Host Disease (GvHD) and for Host-Versus-Graft Disease(HvGD) that will compromise effectiveness and duration of treatment. Knockout of the endogenous T cell receptor and alteration of major histocompatibility complex class I and class II molecules of the CAR-T cells are gene editing strategies that are being adopted to limit GvHD and HvGD.17,18 Gene editing has also been used to ablate CD52 and make the infused cells resistant to an anti-CD52 lymphodepleting agent, alemtuzumab, which can be used to promote engraftment of CAR-T cells.19 Aside from genetic manipulations required to generate safe allogeneic CAR-T cells, additional editing is likely needed to make CAR-T cells efficient against solid tumors. To date CAR-T cell therapies have fared poorly as treatments for solid tumors potentially owing to the immunosuppressive environment, CAR-T cell exhaustion and consequently reduced persistence of the engrafted cells after infusion. Multiple factors, PD-1, CTLA-4, LAG- 3, and TIM-3 to mention a few, when depleted increase persistence and activity of CAR-T cells.20 However, when multiple genes are targeted using nuclease technologies, such as CRISPR-Cas9, several DSBs are simultaneously generated in the genome with a high risk of deleterious chromosomal abnormalities impacting the safety of the therapy.17,19,21,22 As the number of DSBs increases, so does the number of possible chromosomal translocations increasing the risk that the edited cells will die or sustain potentially pro-tumorigenic mutations, compromising the whole cell therapy strategy. An alternative is to use base editors to knockout genes by introducing premature stop codons or destroying splice sites, offering the possibility of safer multiple gene knockouts without the DSBs.22,23 Both CBEs and ABEs have successfully generated CAR-T cells in which multiple genes are knocked out.12,22 For example, Webber and colleagues used a CBE to knockout B2M, CD52, TRAC and PDCD1 and showed that the cells retained functionality and were able to efficiently kill cancer cells in vitro. Moreover, there was no evidence of chromosomal translocations observed in T cells similarly edited with CRISPR-Cas9 technology.22 Another issue requiring an editing solution is fratricide (the killing of T cells by T cells) when generating CAR-T cells to target T cell leukemia or lymphoma. The CARs in this case are targeting T cell antigens, such as CD3 and CD7, and if these antigens remain expressed on the surface of the engineered CAR-T cells, the cells will target each other during the manufacturing process. Base editing was used to initially knockout TRBC, CD3 and CD7 ahead of introducing a CAR against CD3 or CD7.24 The cells edited with base editors showed significant reduction in chromosomal translocation events compared with the cells edited with conventional CRISPR-Cas9 approaches. These initial studies support the hypothesis that efficient multiplex engineering of therapeutic primary cells is possible with base editors and that the safety profile in the resulting cells is favorable compared with nuclease-based technologies.
Challenges for Therapeutic Base Editing
Base editing has the potential to be a first-class player in the future of cell and gene therapy providing treatment options for cancer and genetic diseases. Addressing targeting scope and safety limitations of the technology is of utmost importance for the success of base editing in the therapeutics arena. Unwanted off-target modifications in DNA and/or RNA are observed with base editors, raising safety concerns. Some of these concerns are being addressed through the generation of high-precision base editors with reduced or undetectable off-target effects combined with transient delivery modalities. Currently, the ability to modify a specific base depends on its relative position within the target locus specified by the guide RNA and on the availability of an enzyme able to make the required base conversion. The recent publication of a C to G transversion base editor25 and the use of less sequence constrained Cas proteins should aid the wider application of base editors in cell and gene therapy.
The base editing and nuclease gene editing fields are evolving rapidly with successes and challenges evident on both sides. Both are being embraced by the cell and gene therapy communities and both have much to offer patients with genetic conditions. Ultimately, the long- term safety profile of these technologies will likely determine which approach will be used in the gene and cell therapies of the future.
References
- Frangoul, H. et al. (2021) CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 384, 252–260.
- Nishida, K. et al. (2016) Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353.
- Komor, A. C.et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424.
- Gaudelli, N. M. et al. (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471.
- Lavrov, A. V. et al (2020) Genome scale analysis of pathogenic variants targetable for single base editing. BMC Med. Genomics 13, 80 (2020).
- Levy, J. M. et al. (2020) Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 4, 97–110.
- Villiger, L. et al. (2021) In vivo cytidine base editing of hepatocytes without detectable off- target mutations in RNA and DNA. Nat. Biomed. Eng. doi:10.1038/s41551-020-00671-z.
- Ryu, S.-M. et al. (2018) Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 36, 536–539.
- Koblan, L. W. et al. (2021) In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature 589, 608–614.
- Mok, B. Y. et al. (2020) A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637.
- JPM: Verve Therapeutics unveils its lead program—a one-and-done treatment for genetic high cholesterol. FierceBiotech https://www.fiercebiotech.com/biotech/jpm-verve- therapeutics-unveils-its-lead-program-a-one-and-done-treatment-for-genetic-high.
- Gaudelli, N. M. et al. (2020) Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 38, 892–900.
- KYMRIAH (tisagenlecleucel), US Food and Drug Administration, https://www.fda.gov/ vaccines-blood-biologics/cellular-gene-therapy-products/kymriah-tisagenlecleucel
- YESCARTA (axicabtagene ciloleucel), US Food and Drug Administration, https://www.fda. gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene- ciloleucel
- BREYANZI (lisocabtagene maraleucel), US Food and Drug Administration, https:// www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/breyanzi- lisocabtagene-maraleucel
- TECARTUS (brexucabtagene autoleucel), US Food and Drug Administration, https:// www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/tecartus- brexucabtagene-autoleucel
- Qasim, W. et al. (2017) Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9.
- Ren, J. et al. (2017) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 23, 2255–2266.
- Poirot, L. et al. (2015) Multiplex Genome-Edited T-cell Manufacturing Platform for‘Off-the- Shelf’ Adoptive T-cell Immunotherapies. Cancer Res. 75, 3853–3864.
- Pavlovic, K. et al. (2020) Using Gene Editing Approaches to Fine-Tune the Immune System. Front. Immunol. 11, 570672.
- Stadtmauer, E. A. et al. (2020) CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365.
- Webber, B. R. et al. (2019) Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 10, 5222.
- Billon, P. et al. (2017) CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell 67, 1068-1079.e4.
- Georgiadis, C. et al. (2020) Base-edited CAR T Cells for combinational therapy against T cell malignancies. bioRxiv 2020.07.30.228429 doi:10.1101/2020.07.30.228429.
- Kurt, I. C. et al. (2021) CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39, 41–46.
- Geurts, M. H. et al. (2020) CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell 26, 503-510.e7.
- Yeh, W.-H. et al. (2020) In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 12.
- Zeng, J. et al. (2020) Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 26, 535–541.
- Chadwick, A. C. et al. (2017) In Vivo Base Editing of PCSK9 (Proprotein Convertase Subtilisin/ Kexin Type 9) as a Therapeutic Alternative to Genome Editing. Arterioscler. Thromb. Vasc. Biol. 37, 1741–1747.
- Lim, C. K. W. et al. (2020) Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 28, 1177–1189.
Author Biography
Immacolata Porreca, PhD, is a Senior Scientist in the R&D Base Editing team at Horizon Discovery, a PerkinElmer company. Since joining the company in November 2019, she is contributing to the development and commercialization of the base editing platform Pin-point™, with a particular focus on the therapeutic applications of the technology. Before joining Horizon, Immacolata was a PostDoc at the Human Genetic Department of the Wellcome Sanger Institute in Cambridge characterizing human variation in HIV infection and prior to that she investigated endocrine disruption at the Biogem Institute in Italy. Immacolata gained her doctorate in Human Genetic at the University of Naples, Italy. Immacolata.Porreca@ horizondiscovery.com, horizondiscovery.com