Luis Jimenez, Yarah Abazah, Tae Min Kim, Brittany Cardona, Anna Maclejewska, Laila Metwaly, Shakila Behzadi, Amy Kass Georges, Umar Sultan Kahloon, and Hadassah Haricha
Biology and Horticulture Department, Bergen Community College, Paramus, New Jersey Send all correspondence to: Luis Jimenez, Biology and Horticulture Department, Bergen Community College, Paramus, New Jersey, Telephone: 201-446-7143, Email: ljimenez@bergen.edu
Abstract
The COVID-19 pandemic has presented major challenges to national public health systems and created serious socio-economics disruptions around the world. A zoonotic event that triggered the spillover of SARS-CoV-2 to human populations has not been decisively described. However, genomic surveillance of SARS-CoV-2 infections provided rapid viral characterization, rapid diagnostic tests, effective vaccines, and epidemiological analysis of COVID-19. The original virus that emerged out of China has gone through several changes in the nucleic acid sequences of the spike glycoprotein which is the structure used to facilitate entry into human respiratory cells causing morbidity and mortality. These changes triggered the emergence of variants of concerns as a response to evolutionary pressures to optimize viral transmissibility and replication. Continuous surveillance of real-time viral evolution as the pandemic proceeds will optimize public health and infection control practices to minimize the impact of COVID-19.
Microbiology of COVID-19
“We come to this conclusion, that the frequent discovery of microorganisms in traumatic infective disease and the experimental investigations made in connection with them render the parasitic nature of these diseases probable.”
- Robert Koch
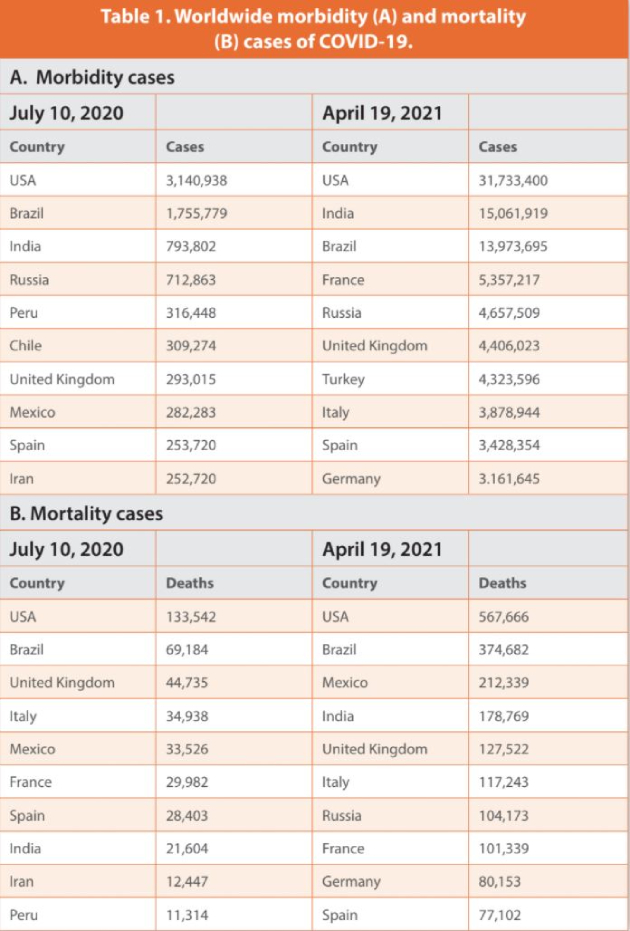
A novel human coronavirus (HCoV), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is the etiological agent responsible for the COVID-19 pandemic. The first cases were reported in Wuhan, China, with a zoonotic event triggered at a wet market by either a spillover from bats or the presence of an intermediary animal host prior to the first human infection.1 Soon after, on January 19, 2020, a Washington state resident became the first case in the USA.2 The patient returned from an overseas trip and presented similar signs and symptoms related to COVID-19. The infection was confirmed by a positive reaction from nasopharyngeal samples using reverse transcription polymerase reaction (RT-PCR). The first person to person transmission in the USA was reported on January 30, 2020 in Illinois.3 Ever since then, morbidity and mortality cases due to COVID-19 have exponentially grown to the point that on April 19, 2021, more than 31 million cases and 567,666 deaths have been reported in the USA (Table 1). After more than a year, the USA has the most cases followed by India, Brazil, France, and Russia. When it comes to mortality USA deaths are 1.5 times (x) more than the second country, e.g., Brazil, followed by Mexico, India, and the United Kingdom (UK). The USA has been the country with the highest numbers of cases and deaths for every month since May 2020 to May 2021 (Figures 1A and 1B). The exponential growth in cases and deaths has been an indication of the insufficient public health practices to control COVID-19. India shows the fastest growth in COVID-19 cases with almost 15x more cases than 10 months ago, followed by Brazil (13x) and the USA (10x) (Table 1). Mortality growth rates are again led by India (8x), Mexico (7x), and Brazil (5x).
Based upon genomic analysis, SARS-CoV-2 appeared to have strong structural and genetic similarities to other RNA viruses such as SARS-CoV and bat coronaviruses (BtCoV).1 SARS-CoV-2 like SARS-CoV and Middle East Respiratory Syndrome Coronavirus (MERS-CoV) belong to the genus Betacoronavirus.1 Although SARS-CoV-2 widespread transmission and infection seem to be much faster than SARS-CoV and MERS-CoV, mortality rates are lower (Table 2). However, MERS-CoV appeared to be less transmissible than SARS-CoV.4 SARS-CoV was found to be transmitted by super-spreader events while MERS-CoV depended on cluster outbreaks.4 SARS-CoV-2 has been widely disseminated by several super spreading events. For SARS-CoV and MERS-CoV person to person transmission was only happening when illness was apparent. SARS and MERS cases put together do not account to more than 11,00 worldwide while COVID-19 cases are more than 141 million (Table 2). This is due to the fact that SARS-CoV-2 is widely disseminated by asymptomatic individuals.1 Approximately 45% of COVID-19 cases are due to asymptomatic transmission. This situation created serious disruptions to the world economy and society in general since asymptomatic individuals can spread the virus without proper testing and contract tracing. Few countries such as China, Australia, Korea, Taiwan, Vietnam, and New Zealand have successfully reduced and controlled COVID-19 by rigorous testing, tracing, and patient isolation. Unfortunately, most countries have not been able to develop proper public health practices to minimize morbidity and mortality cases.
Genomic Surveillance of SARS-CoV-2
Sequencing of viral genomes is a very important tool to understand viral evolution during an outbreak. During the SARS and MERS pandemics, genomic surveillance led to a better understanding of the different outbreaks and contributed to rapid diagnostic tests.5,6 However, the number of viral genomes sequenced from clinical samples between outbreaks were less than 200. Previous studies using genomic sequences from 179 patients infected by the Ebola virus (EbV) in West Africa demonstrated that spike glycoprotein mutations led to higher infectivity rates resulting in significant mortality rates.7 It was the first time in history when genomic information was developed in real-time and used to understand how a virus was transmitted and changing as the epidemic proceeded leading to proactive applications regarding public health and infection control.8
Next generation nucleic acid sequencing, RT-PCR, and bioinformatics have developed new tools that can be deployed to understand viral evolution and transmission when serious zoonotic infections take place. These advances have enabled the rapid whole-genome sequencing of clinical samples from patients with COVID-19 allowing real-time tracing of viral evolution and distribution around the world.9,10 Full genomic sequences of SARS-CoV-2 can be generated within hours or days after samples were found to be positive by RT-PCR.
The first complete genomic sequence of SARS-CoV-2 (Wuhan-1) was used for rapid virus characterization, development of rapid diagnostic assays, and vaccines. Ever since then more than 700,000 viral genomes have been sequenced. According to the Centers for Disease Control and Prevention (CDC) (https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-emerging-variants.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fcoronavirus%2F2019-ncov%2Fmore%2Fscience-and-research%2Fscientific-brief-emerging-variants.html) genomic surveillance provides a way to identify a variant of concern (VOC) which can be leading to the following outcomes:
- Higher transmissibility - the virus spreads faster from person to person
- Higher pathogenicity - increase risk of chronic disease and death
- Evasion to detection by specific viral diagnostic tests - RT-PCR tests target multiple genes to detect SARS-CoV-2. Any mutation on one of those genes can reduce the sensitivity of the assay increasing the numbers of false negatives
- Decrease efficacy of therapeutic drugs - drugs such as monoclonal antibodies (Mabs) will show reduced efficacy to neutralize the virus
- Evasion to natural or vaccine-induced immunity - variants go undetected by the immune system reducing vaccine effi cacy and protective immune response


Continuous genome sequencing of SARS-CoV-2 variants during the COVID-19 pandemic has provided important information on how the virus is evolving as it goes through different populations worldwide.11 The development of messenger RNA (mRNA) vaccines allows the rapid response to any mutation or variants with the ability to significantly reduce vaccine efficacy to levels where protection will be severely compromised leading to surges in morbidity and mortality.12 These vaccines are based upon new technology that provided the fastest development and deployment in the history of vaccinology. A two dose regimen of the vaccines provided more than 90% protection against COVID-19 in subjects 16 years of age and older.12
SARS-CoV-2 Genome and Proteins Related to Transmission and Infection
"Disease usually represents the inconclusive negotiations for symbiosis... a biological misinterpretation of borders."
- Lewis Thomas
The genome of SARS-CoV-2 consists of single stranded RNA with 29.9 kilobases (kb) which is bigger than SARS-CoV but smaller than MERS- CoV (Table 2). The genome has 14 open reading frames (ORF) coding for 27 proteins.13 Structural proteins are coded by four diff erent genes (1). These proteins are spike glycoprotein (S), envelope (E), membrane (M), and nucleocapsid (N) as well as several accessory proteins that participate in viral replication, infectivity and pathogenesis (Figure 2). The E protein helps with virus assembly and release. It also enhances virus pathogenicity. The M protein is the most abundant protein maintaining viral assembly, integrity, and stability. The N is a nucleocapsid protein essential in the packaging and protection of encapsulated viral genomes. One of the most important accessory proteins is the RNA-dependent RNA polymerase (RdRp) that catalyzes the synthesis of new viral RNA.

However, it’s S that binds human angiotensin converting enzyme-2 (ACE2) to promote viral attachment and entry to the host cell.14 The spike has two Subunits named 1 and 2 (Figure 3).1,14 The spike is a trimeric protein consisting of 1273 amino acids coded by a gene with a size of 3,822 bases (Figure 4). Subunit 1 (Sb1) contains 672 amino acids (residues 14 to 685)(Figure 4). The Sb1 also contains the receptor binding domain (RBD) which is attaching ACE2 (Figures 3 and 4). Subunit 2 (Sb2) is a transmembrane subunit promoting fusion of the host and viral cell membranes and entry by endocytosis. The Sb2 is composed of 588 amino acids (residues 686 to 1273)(Figure 4). Although SARS-CoV-2 genomic comparison studies showed high similarities to bat coronaviruses, 96% similarity, the S gene was closer to SARS-CoV than any other coronavirus. Recent studies showed the spike of pangolin CoVs with an even closer genetic similarity to SARS-CoV-2 even though genome relatedness was distant.14
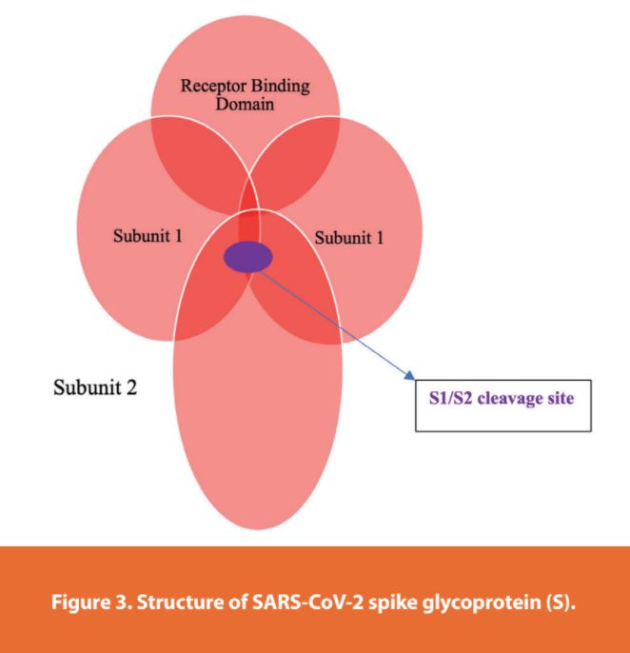

However, the spike of SARS-CoV-2 showed more affi nity and stability to ACE2 than SARS-CoV. There is also a cleavage site at the junction of the Sb1 and Sb2 subunits (Figures 3 and 4). This cleavage site is not found in any other bat or pangolin coronavirus. Entry to cells is enhanced by a host protease, transmembrane protease serine 2 (TMPRSS2). Upon entry, viral envelope fusion with cell membrane through the endosomal pathway leads to the releasing of viral RNA into the host cell.
SARS-CoV-2 Variants of Concern
"This preservation of favorable variations and the destruction of injurious variations, I call Natural Selection, or the Survival of the Fittest. Variations neither useful nor injurious would not be affected by natural selection and would be left a fluctuating element."
- Charles Darwin
Coronaviruses infecting bats are adapted to fast metabolic rates which are necessary for volant mammals.15,16 Unique antiviral defenses systems in bats such as continuous interferon response and specific anti-inflammatory traits keep BtCoV under control. BtCoV countered these adaptations by rapid spreading from cell to cell without creating significant pathologies within the host. Therefore, faster transmission rates among other things are basically how BtCoV adapted to vigorous immunological responses in bats. However, these immune responses are absent in non-volant mammals and when viruses spilled into human or other animal populations serious zoonoses such as rabies, Hendra, Nipah, Marburg, SARS, MERS, Ebola and COVID-19 can occur.
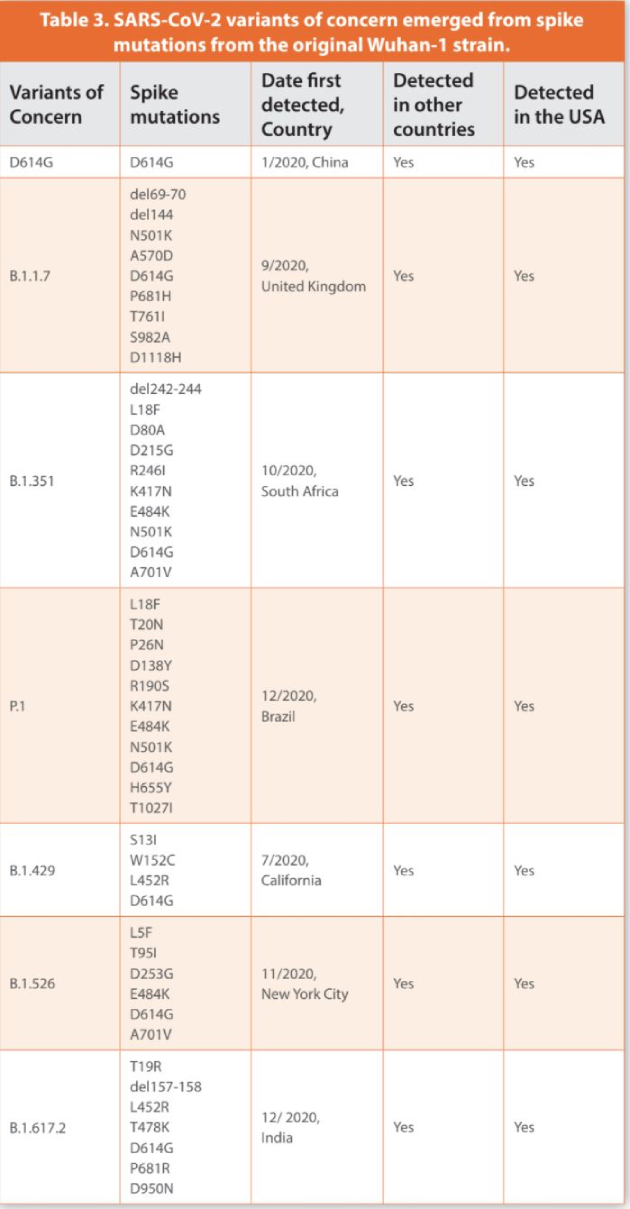
The original D614 (Wuhan-1) strain of SARS-CoV-2 which was disseminated from China to the world mutated into variant D614G (Table 3).17 After analyzing more than 48,000 SARS-CoV-2 genome samples, the most prevalent mutation was found to be in the S protein.11 The D614G mutation took place in position 614 with a substitution of the amino acid aspartic acid by glycine as a result of an A to G nucleotide mutation (Figure 4). This mutation was located outside the RBD region of S but was present at the intersection between Sb1 and Sb2 (Figure 4). However, it triggered conformational changes increasing the affinity of S to ACE2. Fortunately, this change did not make this variant more lethal. Furthermore, it did not interfere with antibody neutralization nor reduced vaccine efficacy.18 Variation analysis in early March of 2020 found that D614G was not widely distributed globally but was detected in Europe in higher frequencies than in China.17 However, by June 20, 2020, it was the predominant pandemic strain of COVID-19. The new variant was related to higher viral loads and infectivity.
Because SARS-CoV-2 is adapting to a new human host, other mutations are emerging in different regions of S. Changes in the genetic sequences of S and other SARS-CoV-2 genes are triggered by evolutionary pressure on the virus to adapt to a new host in order to optimize survival and replication.19 When SARS-CoV-2 replicates in human cells, thousands of mutations are taking place. These mutations can be either neutral, harmful, or beneficial to viral fitness. Most mutations are inconsequential. However, over time they build up on top of each other and can lead to new variants. Some mutations can reduce the efficacy of Mabs and vaccines by allowing immune escape.20,21 SARS and MERS, are more lethal than SARS-CoV-2. However, they showed lower transmission rates as shown in Table 2. Because their outbreaks were rapidly controlled and case numbers were kept significantly lower than COVID-19 there was no evolutionary pressure, compared to SARS-CoV-2, to develop as many new variants with different mutations.
Most mutations in SARS-CoV-2 are single changes of amino acids called substitutions(Figure 4).11 Although SARS-CoV-2 and other HCoV possess proofreading abilities, an exoribonuclease enzyme (ExoN) to correct genetic changes, natural selection drives the development of variants enhancing SARS-CoV-2 fitness to be more transmissible and infectious. Furthermore, in some cases changes are driven by deletions (del) in S (Figure 4). These deletions cannot be corrected by ExoN enzymes.20 For instance, influenza viruses (IV) undergo similar genetic changes based upon the gradual accumulation of random mutations or antigenic shift during the flu season.22 However, IV do not have proofreading abilities like HCoV. Therefore, mutation rates are extremely high when compared to HCoV. Nevertheless, the longer COVID-19 goes on the more chances SARS-CoV-2 will have to accumulate relevant mutations in S which may compromise vaccine efficacy and drug treatment. All vaccines and monoclonal Mabs treatments developed so far are targeting S. Therefore, SARS-CoV-2 is under evolutionary pressure to develop variants that can overcome treatments developed to control the pandemic. While changes in S are important other mutations in different SARS-CoV-2 genes contribute to enhanced viral transmission and disease severity. Peacock et al.23 discussed several important genetic mutations taking place in proteins such as RdRp and N. However, in this article we will limit our discussion only to mutations in S.
Three months after D614G became the dominant pandemic variant, a VOC was detected in the UK. The VOC named B.1.1.7 accounted for most COVID-19 cases in the UK (Table 3).24 Higher viral loads were detected in COVID-19 patients. In addition to the D614G mutation, this variant showed, for the first time, eight amino acid changes within S (Table 3). One of the changes was replacing the amino acid asparagine for tyrosine in position 501, mutation N501Y (Figure 4). This particular mutation located in the RBD region of S enhanced the affinity and binding to ACE2 making B.1.1.7 more transmissible and causing more severe disease.25 Similar mutations have been observed in the RBD of the IV spike protein allowing immune escape.22
B.1.1.7 also showed two deletions found in positions 69 and 144 of Sb1 which were not previously reported for variant D614G (Figure 4). These deletions enabled the virus to infect cells more efficiently. Another mutation was detected nearby the cleavage site of S, P681H. Several other substitutions not previously reported in any VOC such as T761I, S982A and D1118H were detected in Sb2 (Figure 4). According to the CDC, on April 9, 2021, B.1.1.7 became the most common variant causing COVID-19 cases in the USA leading to a surge in morbidity and mortality cases across the nation (https://www.cdc.gov/coronavirus/2019-ncov/transmission/variant-cases.html). Several states such as California, Michigan and Florida were severely affected by this VOC. While COVID-19 cases are expected to increase nationwide due to infections with B.1.1.7, hospitalizations and deaths will likely remain at lower levels where high vaccination coverage and proper safety measures are implemented.
Another variant independently emerged in South Africa named B.1.351. B.1.351 shows 10 mutations in S (Table 3). Like previous VOC it carries the D614G and N501Y mutations (Figure 4). However, it shows a unique change in position 484 where the amino acid glutamic acid was replaced with lysine, mutation E484K.26 This mutation took place in the RBD region of S (Figure 4). The RBD region of B.1.351 showed a second novel mutation, K417N, a lysine to asparagine substitution. B.1.351 was the first VOC emerging with 3 mutations in the RBD region. New substitutions and deletions were detected in B.1.351 mostly located in Sb1 (L18F, D80A, D215G, R246I, del242-244). However, only one mutation was detected in Sb2 (A701V). Vaccine and Mabs efficacy against B.1.351 was reported to be lower than previous VOC indicating that mutation E484K might be compromising immune response.18 Furthermore, studies demonstrated B.1.351 to be more transmissible and virulent than previous VOC.25
Unfortunately, the same E484K mutation was found in a new VOC, P.1, detected in Brazil and Japan.27 P.1 has eleven changes in S (Table 3). Several cases of reinfection in the city of Manaus, Brazil, demonstrated that P.1 initiated a new wave of morbidity and mortality. This variant shared RBD mutations such as K417N and N501Y with other VOC (Figure 4). P.1 also shared mutations L18F and D614G with B.1.1351 There were six new mutations in S that were not previously reported but emerged in P.1. They were T20N, P26N, D138Y, R190S in Sb1, one nearby the cleavage site, H655Y, and one in Sb2, T1027I (Figure 4). All these changes either enhanced virus transmission, immune evasion, or their relevance is currently unknown.
A surge in COVID-19 cases in Southern California (SC) since October 2020 was driven by variant B.1.429.28 Three distinctive mutations were found with substitution L452R located in the RBD and two new substitutions in Sb1, S13I, and W152C (Table 3). Based upon genomic analysis more than 40% of COVID-19 cases in SC were due to this variant. The SC variant shows the D614G mutation but did not have other RBD substitutions previously detected (K417N, E484K, N501Y) (Figure 4). Compared to previous VOC, no mutations were detected in Sb2. Based upon clinical data, B.1.429 appeared to be more infectious than other variants found in SC and caused more severe illness.28 By March 31, 2021, B.1.429 was responsible for 67% of COVID-19 infections in Colorado.29 Several cases were reported where Mabs efficacy against this VOC was significantly reduced.
A new VOC also emerged in New York (NY) city named B.1.526. This VOC showed 3 new mutations in Sb1 (Table 3). They were L5F, T95I, D253G. However, B.1.526 shared substitutions E484K and D614G with other VOC (Figure 4). There was a substitution in Sb2 where the amino acid alanine was replaced by valine, A701V. This mutation was previously reported in B.1.351. Over time COVID-19 cases due to infections with B.1.526 significantly increased in NY city and NY state.30 However, infections with B.1.526 did not lead to more severe disease nor were associated with increased risk for infection after vaccination.
In December 2020, an increase in the number of COVID-19 cases in India were associated with the emergence of a new VOC named B.1.617.2.31 By late March this new VOC accounted for 50% of the cases in the country. Based upon the numbers of new cases driven by B.1.617.2 in the UK, Public Health England (PHE) reported this VOC to be as transmissible as B.1.1.7. It was also reported to be the number one VOC in new cases in the UK and showed the highest growth of all VOC in the USA by the end of June 2021.31,32 B.1.617.2 accounted for more than 30% of COVID-19 cases in several midwestern states in the USA that unfortunately showed very low vaccination rates compared to other areas of the country.
As all other VOC, B.1.617.2 showed the D614G mutation that increases transmissibility and viral load (Figure 4). B.1.617.2 also has the L452R mutation in the RBD along with a unique mutation, T478K, never seen in any other VOC. The L452R mutation, previously reported in the southern California strain, B.1.429, enhanced virus transmissibility and facilitates immune escape. The P681R mutation nearby the cleavage site may enhance cell entry and infectivity (Figure 4). Other unique mutations were D950N in the Sb2 and T19R and del157-158 in the Sb1 region of the S protein.
In conclusion, the most abundant S mutation present in all VOC is the D614G substitution (Figure 4)(Table 3). This mutation enhanced binding to ACE2, virus transmissibility and replication. Some mutations in the RBD such as K417N, E484K, and N501Y were present in half of VOC but emerged later in the pandemic as a response to evolutionary pressure to optimize viral transmissibility and replication (Figure 4) (Table 3). These changes led to immune escape and stronger binding to ACE2. In general, most mutations seemed to be happening in Sb1 with multiple deletions and substitutions while Sb2 might not be encountering similar evolutionary pressure. Because of the emergence of VOC, due to natural selection, showing increased immune escape, transmissibility, and infectivity, continuous genomic surveillance of SARS-CoV-2 evolution will lead to better management of COVID-19. Rapid upgrade of current vaccines when needed will provide vigorous immunity to any emerging VOC. However, although efficacious vaccines have been developed, the COVID-19 pandemic is far from over. The uneven deployment and distributions of effective vaccines around the world do not support the common goal of eliminating COVID-19. In some countries new cases are driven by VOC emerging or replacing previous ones as a response to evolutionary pressure on SARS-CoV-2. A major international commitment with proper coordination and efficient vaccine deployment along with genomic surveillance of SARS-CoV-2 will be needed to manage and reduce the impact of COVID-19 on global public health and socioeconomics structures around the world.
References
- Rabaan, A.A. et al. 2020. SARS-CoV-2, SARS-CoV, and MERS-CoV: A comparative overview. Le Infezioni in Medicina 2:174-184.
- Holshue, M.L. et al. 2020. First case of 2019 novel coronavirus in the United States. New England Journal of Medicine 382:929-936.
- Ghinai, I. et al. 2020. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet 395:1137-1144.
- Wong, G. et al. 2015. MERS, SARS, and Ebola: The role of super-spreaders in infectious disease. Cell Host & Microbes 18:398-401.
- Guan, Y. et al. 2004. Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome. Lancet 363:99-104.
- Cotton, M. et al. 2013. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet 382:1993-2002.
- Diehl, W.E. et al. 2016. Ebola virus glycoprotein with increased infectivity dominated the 2013-2016 epidemic. Cell 167:1088-1098.
- Holmes, E.C. et al. 2016. The evolution of Ebola virus: Insights from the 2013-2016 epidemic. Nature 538:193-200.
- Pereson, M.J. et al. 2020. Phylogenetic analysis of SARS-CoV-2 in the first few months since its emergence. Journal of Medical Virology 93:1722-1731.
- Lu, R. et al. 2020. Genomic characterization and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395:565–574.
- Mercatelli, D. and Giorgi, F.M. 2020. Geographic and genomic distribution of SARS-CoV-2 mutations. Frontiers in Microbiology 11: 1800. doi:10.3389/fmicb.2020.01800.
- Polack, F.P. et al. 2021. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. New England Journal of Medicine 383:2603-2615.
- Jimenez, L. et al. 2020. Microbiology of COVID-19: Chronicle of an announced pandemic. American Pharmaceutical Review 23: 140-147.
- He, J. et al. 2020. Molecular mechanism of evolution and human infection with SARS-CoV-2. Viruses 12:428; doi:10.3390/v12040428.
- Brook, C.E. et al. 2020. Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. eLife 2020;9:e48401. DOI: https://doi.org/10.7554/eLife.48401.
- ebb, D. et al. 2020. Six reference-quality genomes reveal evolution of bat adaptations. Nature 583:578-584.
- Korber, B. et al. 2020. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 182:812-827.
- Planas, D. et al. 2021. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B1.351 variants to neutralizing antibodies. Nature Medicine. https://doi.org/10.1038/s41591-021-01318-5.
- Duan, L. et al. 2020. SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Frontiers in Immunology https://doi.org/10.3389/fimmu.2020.576622.
- McCarthy, K.R. et al. 2021. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 371:1139-1142.
- Weisblum, Y. et al. 2020. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020;9:e61312. DOI: https://doi.org/10.7554/eLife.61312 .
- Fonville, J.M. et al. 2014. Antibody landscapes after influenza virus infection or vaccination. Science 346:996-1000.
- Peacock, T.P. et al. 2021. SARS-CoV-2 one year on: evidence for ongoing viral adaptation. Journal of General Virology:102:001584 DOI 10.1099/jgv.0.001584
- Frampton, D. et al. 2021. Genomic characteristics and clinical effect of the emergent SARS- CoV-2 B.1.1.7 lineage in London, UK: a whole-genome sequencing and hospital-based cohort study. Lancet https://doi.org/10.1016/ S1473-3099(21)00170-5.
- Khan, A. et al. 2021. Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data. Journal of Cell Physiology DOI:10.1002/jcp.30367.
- Planas, D. et al. 2021. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B1.351 variants to neutralizing antibodies. Nature Medicine. https://doi.org/10.1038/s41591-021-01318-5.
- Sabino, E.C. et al. 2021. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. https://doi.org/10.1016/ S0140-6736(21)00183-5.
- Zhang, W. et al. 2021. Emergence of a novel SARS-CoV-2 variant in Southern California. Journal of the American Medical Association 325:1324-1326.
- 29. Webb, L.M. et al. 2021. Identification of and surveillance for the SARS-CoV-2 variants B.1.427 and B.1.429-Colorado, January-March 2021. Morbidity and Mortality Weekly Reports ePuB: 5 May 2021. DOI: http://dx.doi.org/10.15585/mmwr.mm7019e2.
- 30. Thompson, C.N. et al. 2021. Rapid emergence and epidemiological characteristics of the SARS-CoV-2 B.1.526 variant-New York city, New York, January 1-April 5, 202. Morbidity and Mortality Weekly Reports ePuB: 5 May 2021. DOI: http://dx.doi.org/10.15585/mmwr.mm7019e1.
- European Centre for Disease Prevention and Control. 2021. Emergence of SARS-CoV-2 B.1.617 variants in India and situation in the EU/EEA-11 May 2021. ECDC: Stockholm, 2021.
- Centers for Disease Control and Prevention. 2021. https://covid.cdc.gov/covid-data-tracker/#variant-proportions. May 29,
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!