By: Asha Hewarathna,a,§ Cynthia Sommers,a Barry Rosenzweig,b Jason Rodriguez,a David Keire,c and Kui Yanga*
- a: Division of Complex Drug Analysis, Office of Testing and Research, Center for Drug Evaluationand Research, U.S. Food and Drug Administration, St. Louis, MO.
- b: Division of Applied Regulatory Science, Office of Clinical Pharmacology, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD.
- c: Office of Testing and Research, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, St. Louis, MO.
- §: Currently with Division of Biotechnology Review and Research III, Office of Biotechnology Products, Center for Drug Evaluation and Research, U.S. Food and Drug Administration, Silver Spring, MD.
- *: Corresponding author. Email address: Kui.Yang@fda.hhs.gov
Abstract
Protamine Sulfate (ProS) is the only FDA approved anticoagulation reversal agent for unfractionated heparin. ProS is biologically derived from chum salmon sperm and therefore may be contaminated with residual salmon sperm DNA. Because residual host cell DNA (resDNA) may pose a safety concern to patients, the amount should be closely monitored during manufacturing and strictly regulated in final drug products. The regulatorily defined acceptance level (10 ng per dose) of resDNA in drug products poses great challenges for accurate quantification of resDNA, particularly for the products used in high doses or formulated in complex matrixes. resDNA quantification in the presence of the cationic arginine-rich ProS structure can be challenging due to the polyanionic resDNA binding to ProS which may render isolation and analysis of resDNA problematic. In this work, a droplet digital polymerase chain reaction (ddPCR) method was adapted to quantify resDNA in ProS drug product. The ddPCR reaction was tolerant to matrix interferences thus eliminating the need for DNA extraction required with traditional PCR assay. In addition, the method provided absolute DNA quantification without the need for calibration curves. The developed ddPCR assay had a lower limit of quantitation (LLOQ) of 1.67 pg of DNA per mg of ProS (1.67 ppb) or 83 pg per 50 mg dose of ProS, a wide dynamic range spanning three orders of magnitude and an average % recovery of 82.5% with inter-day coefficient of variations (CV) < 20% across the entire concentration range. Overall, the ddPCR analytical procedure replaced the need for comprehensive DNA extraction with a simple in-plate protease digestion of ProS and was 120-fold more sensitive than the acceptable amount of resDNA. Five lots of ProS drug products were analyzed using this method and none tested positive for resDNA contamination above the method’s LLOQ. The method may also be promising for quantifying resDNA in other challenging protein drug products which have a propensity to bind DNA or have other matrix-related complexities.
Keywords: Protamine sulfate, Digital droplet polymerase chain reaction (ddPCR), Residual host cell DNA, DNA quantification
FDA Disclaimer: The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Introduction
The use of biopharmaceuticals has increased in recent years due to their superior efficacy and safety in targeted treatments in comparison to small-molecule drugs.1 Biopharmaceuticals have become one of the most effective clinical treatments for a broad spectrum of diseases including cancers, metabolic disorders and autoimmune diseases.2,3 Biopharmaceuticals are biologically sourced drugs derived from living cells, including those produced from cell lines (e.g., bacterial, yeast or mammalian cell) or manufactured by purification from animal or plant sources. Biopharmaceuticals, therefore, may be potentially contaminated with process-related host cell impurities including residual nucleic acids (DNA and RNA), proteins or lipids from the host organism.4,5 The host cell-originating DNA molecules, referred to as residual host cell DNA (resDNA), may pose potential risks of immunogenicity, infectivity, oncogenicity or mutagenesis to patients.6,7 Residual DNA is therefore closely monitored during manufacturing to assure its adequate clearance, and in the final drug product to ensure the amounts are within acceptable levels. The U.S. Food and Drug Administration (FDA) and World Health Organization (WHO) recommend an acceptable limit of ≤10 ng of residual DNA with the size of ≤200 base pairs per dose of drug product, but also suggest a risk assessment to define the DNA upper limit for a particular product or application based on multiple parameters including the biological nature of the host cell, inactivation process and size distribution of the DNA fragments.8
Protamine Sulfate (ProS) is a biologically derived drug product that is routinely used to neutralize heparin during surgical procedures, and the only FDA approved reversal agent for unfractionated heparin.9-11 ProS is an arginine-rich, highly positively charged polypeptide that, when exposed to strongly anionic unfractionated heparin, forms an inactive salt aggregate and reverses the heparin-induced anticoagulation.12,13 The same structural elements also provide ProS a unique DNA binding domain.14,15 For example, ProS can bind and precipitate DNA through electrostatic interactions between cationic arginine residues and anionic phosphodiester groups of DNA.16,17 Because ProS is biologically sourced (primarily isolated from chum salmon sperm),18 some resDNA may still be present even after the purification process due to binding with cationic ProS. Thus, resDNA in ProS should be accurately measured and controlled to meet safety guidelines.
Accurate and sensitive quantification of resDNA impurities in biopharmaceuticals is typically challenging due to inherent low-levels and the potential for interference from inhibitor-rich sample matrices including high-level active product ingredients and excipients. The DNA binding nature of ProS, however, presents an additional unique challenge to resDNA analysis in ProS intermediates and final products. For example, by binding to anionic DNA, cationic ProS may strongly interfere with the DNA assay if unremoved, or greatly reduce resDNA recovery during DNA extraction intended to remove matrix interferences. Quantitative real-time polymerase chain reaction (qPCR) is a well-known DNA analysis technique that has been widely used for the detection and quantification of resDNA in the biotechnology industry due to its broad dynamic range and high sensitivity.7,19-23 Recently we reported a qPCR method that, in combination with an extraction procedure, achieved accurate resDNA quantification in ProS drug products with a demonstrated sensitivity 40-fold higher than the current guidelines for resDNA (10 ng DNA per dose).15,24 qPCR measures the amplification rate of a DNA target in a test sample and quantifies the DNA concentration using a calibration curve of DNA standards.25,26 Because qPCR relies on the accurate measurement of a dynamic DNA amplification and use of high quality DNA standards, intrinsic fluctuations may be present within‐ or between‐labs leading to poor reproducibility and precision. The lack of compendial DNA standards may also add to the overall variability in addition to inter-lab technical variability.24 Recently, droplet digital PCR (ddPCR) technology has become available enabling independent quantification of DNA targets without calibration curves, providing improved reproducibility and precision over qPCR.27-31 In ddPCR, a test sample is partitioned into many thousands of nanoliter-sized droplets where the template DNA molecules separately reside. Within each individual droplet, discrete PCR amplification of the DNA template is run to its end-point, meaning that a single sample now generates tens of thousands of data points rather than a single result. At the end-point, each droplet is analyzed individually and assigned as positive or negative (i.e., the presence or absence of the intended DNA target) by fluorescence analysis and quantified using Poisson statistics. Overall, the benefits of ddPCR technology include, but are not limited to, (1) providing an absolute quantification of target DNA copies; (2) offering great precision enabling the detection of small fold differences; (3) increasing tolerance to interfering substances; and (4) removing PCR bias and improving reproducibility by removing the amplification efficiency reliance of qPCR.30,32,33
The benefits of ddPCR technology, predominantly its improved tolerance to interferences have greatly promoted its recent applications in resDNA analysis in biopharmaceuticals produced from various biological sources including CHO, E. Coli and yeast.22,34-37 In this study, we examined the feasibility of using ddPCR to address the unique challenges in resDNA analysis in ProS samples. In comparison to our qPCR method for ProS,15 the developed ddPCR method is 3 fold more sensitive and does not require DNA extraction (Table 1). All five lots of ProS drug products analyzed using this method had resDNA levels below the method’s LLOQ.
Materials and Methods
Materials
Automated Droplet Generator (ADG), QX200 Droplet Reader, PX1 PCR Plate Sealer and C1000 Touch Thermal Cycler were purchased from Bio-Rad (Hercule, CA). Sterile TE buffer (10 mM Tris, 1 mM EDTA, pH=8), 20% SDS and Proteinase K (~ 20 mg/mL) were purchased from Thermo Fisher Scientific (Dallas, TX). ddPCR Supermix for probes (no dUTP), QX200 droplet generation oil for probes, ddPCR Droplet Reader oil, DG32 Cartridges, ddPCR 96-well PCR plates, Pierceable heat-seal foils, pipet tips for the ADG were purchased from Bio-Rad (Hercule, CA). PCR grade pipet tips were purchased from Eppendorf North America (Hauppauge, NY).
The ddPCR assay targets a conserved region of the multicopy gene (5S rDNA) in the chum salmon genome to achieve maximal sensitivity. The design of TaqMan primers and probes for this target sequence was described previously.24 The primers (forward primer: 5’-AGG GTC GGG CCT GGT TAG-3’, reverse primer: 5’-AAA GCT TAC AGC ACC TGG TAT TCC-3’) and TaqMan probe (5’-ACT TGG ATG GGA GAC CG- 3’) were custom-synthesized at Life Technologies (Grand Island, NY). A commercially available and highly characterized DNA reference material (Ultrapure salmon sperm DNA, Cat #15,632,011) was purchased from ThermoFisher Scientific (Dallas, TX) and used as the DNA standard in this work.
Protamine Sulfate drug products (10 mg/mL for injection, supplied by Fresenius Kabi, USA) were purchased through Bradley Drugs (Bethesda, MD). Five lots were used: 6,110,173 (Lot 1; exp 08/2016), 6,112,326 (Lot 2; exp 07/2017), 6,112,328 (Lot 3; exp 07/2017), 6,112,530 (Lot 4; exp 08/2017), and 6,112,327 (Lot 5; exp 07/2017).
Drug product pretreatment and ddPCR assay
In the ddPCR platform (Bio-Rad), 20 μL of a PCR reaction mixture containing DNA, primers, probes and ddPCR Supermix was transferred for generation of droplets, followed by PCR amplification and droplet analysis. To ensure an accurate transferring of 20 μL of reaction mixture, an initial reaction volume of slightly > 20 μL was prepared, typically a 22 μL containing 12 μL of MasterMix (combined primers, probes and Supermix) and 10 μL of sample solution.
A primer/probe mix was first prepared containing 18 μM primers and 5 μM probe in sterile water. A MasterMix was prepared containing 33 μL of primer/probe mix, 330 μL of ddPCR Supermix for probes (no dUTP) and 147 μL of sterile water per 40 assay wells (e.g., up to 10 samples with 4 replicates per sample, or 8 samples with 5 replicates per sample). Twelve μL of MasterMix was added to each well, followed by addition of 10 μL of TE buffer (no-template control, NTC), or 10 μL of samples after digestion including DNA alone (blank controls, for constructing calibration curve), DNA spike-in ProS (for measuring % recovery of spiked-in DNA from sample matrices) or ProS alone (for measuring residual DNA in the drug product).
Sample digestion was carried out on a ddPCR 96-well plate prior to ddPCR analysis. For samples containing ProS, a reaction mixture containing 15 μL of ProS drug product (150 μg of ProS), 10 μL of 2% SDS, 5 μL of Proteinase K (~ 20 mg/mL) and 60 μL of TE buffer was prepared. Ten μL of TE buffer (ProS alone samples) or salmon sperm DNA solution (spike-in ProS samples) with DNA concentrations ranging from 12.5 – 25,000 fg/μL was then added for a final volume of 100 μL and a 10-fold dilution of the reaction components (Tables 1 and 2). For samples containing DNA only (blank controls), a reaction mixture containing 10 μL of 2% SDS and 80 μL of TE buffer was prepared, followed by addition of 10 μL of TE buffer (NTC) or salmon sperm DNA solution (e.g., 125 – 250,000 fg/μL). Next, the plate was sealed with a pierceable heat-seal foil using the Plate Sealer. The protease digestion was carried out at 60°C for 1 hr in the thermal cycler, followed by heat denaturation at 90°C for 15 min.
After digestion, the plate was cooled down to room temperature. For ProS-containing samples, 10 μL of digested solutions (DNA concentration ranges of 1.25 – 2,500 fg/μL or 12.5 – 25,000 fg per reaction) were each transferred into a well containing 12 μL of MasterMix. For ProS-free samples (blank controls), the digested solutions (DNA concentrations ranging from 12.5 – 25,000 fg/μL and SDS concentration of 0.2%) were diluted 10-fold using TE buffer, and 10 μL of each diluted solution was transferred into a well containing 12 μL of MasterMix (Tables 1 and 2). The ddPCR plate was heat-sealed and briefly vortexed, and then supplied to ADG to generate droplets according to the manufacturer’s protocol. The new plate containing the prepared droplets was removed from ADG, heat-sealed and placed in the thermal cycler for PCR reaction. The thermal cycling conditions were set as follows: 10 min at 95°C (initial denaturation), followed by 30 s at 95°C (denaturation) and 60 s at 59.4°C (annealing) with 2°C/s ramp rate for 39 cycles; then 10 min at 98°C (incubation) and held at 12°C infinite (storage).
After the PCR reaction, the plate was loaded to the QX200 Droplet Reader for droplet analysis. Droplets of each well were analyzed sequentially, and fluorescent signals of each droplet were measured individually. The data was analyzed using software QuantaSoft 1.7.4 and QuantaSoft Analysis Pro 1.0 provided with QX200 Droplet Reader. Positive and negative droplets were divided by applying an auto or manual fluorescence amplitude threshold between them. The copy number concentration (copies/μL) of each sample was reported by the ddPCR software to provide absolute quantification of target DNA in the sample.
Thermal gradient optimization in ddPCR assay
To optimize the annealing temperature of the ddPCR reaction, the temperature gradient was performed in the thermal cycler using its inbuilt function to achieve a thermal gradient between 64.4°C and 51.4°C among selected wells, whereas the amount of salmon sperm DNA and primers/probes was fixed per well.
Calibration curve calculations
In ddPCR, a calibration curve is not needed for absolute quantification of target DNA in a sample represented by copy number concentration (copies/μL). However, an established calibration curve (e.g., a linear regression y = a*x + b) is used to convert the measured copies/μL (y) to the weight of DNA in fg (x) in DNA spike-in samples to evaluate assay accuracy or in an unknown sample to report resDNA amount in weight. To generate a calibration curve, the measured copies/μL is plotted against the spiked DNA amount (DNA concentration (fg/μL) or DNA spike per reaction (fg)). Calibration curve statistics are obtained from the linear regression (y = a*x + b) of the data points using, for example, the Microsoft Excel statistics tool.
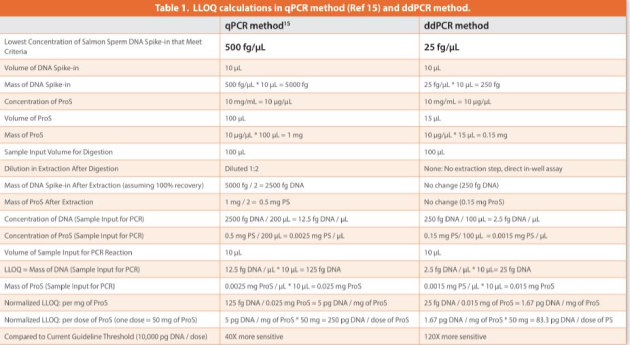
The spiked DNA amount is typically across a few orders of magnitude to achieve a wide dynamic range. A seven-point curve was generated comprising concentration points at 2.5, 5, 10, 20, 100, 500 and 2,500 fg/μL, where closer points were set at the low concentration end (2- fold increases in concentrations up to 20 fg/μL and 5-fold increases thereafter) to emphasize low concentrations where residual DNA measurements most likely reside. A linear regression for such a wide range of concentrations (x-axis), however, inequivalently weighs the data values by overlooking the low concentration data (for example, the four data points from 2.5 to 20 fg/μL shown in Figure 1) while heavily depending on the high concentration data (particularly the data point at the highest concentration 2,500 fg/μL). This bias has a significant negative impact on the assay accuracy at the low concentration end.
A log-log scale is frequently used to resolve this unbalance on x-axis values spanning a wide range. However, from the linear formula y = a*x + b, there is no linear relationship existing between log (x) and log (y). Instead, a linear plot of log (y – b) vs log (x) can be derived from y = a*x + b as follows:
Y = X + c
where Y = log (y – b); X = log (x); c is a constant equal to log (a) and is the y-intercept of the linear plot Y vs X. For this linear plot Y vs X to be a valid derivative from the y = a*x + b, its slope equal to 1 is required. The linearity between y and x (y = a*x + b) is therefore fitted in the plot of Y vs X through iterative change of the constant b until the slope of 1 is yielded (Figure 1), while the simultaneously yielded y-intercept c is used to calculate a (a = 10^c).38 As clearly shown in a log-log scale of these data points (Figure 1C), the linear trendline from Figure 1A, despite the high correlation coefficient (R2 = 0.9998), failed to fit the data points at the low concentration end, while the linear trendline calculated from the linear regression from Figure 1B traced the entire data range fairly well. The approach (demonstrated in Figure 1B) was used in calculation of calibration curve statistics in this study.
The assay LLOQ was determined as the lowest amount of spiked DNA in the established calibration curve with acceptable accuracy and precision. The accuracy (spike recovery) acceptance criterion is set at 50% – 150% of the spiked target DNA according to the current USP <1130> (Residual DNA Testing) Chapter.39 The precision (% CV) acceptance criterion is not specified in the USP <1130> guidance. Up to 36-54% CVs in reported ddPCR assays were generally considered reasonable for low levels of spiked DNA.34-36 The acceptable % CV criterion in this work is set at ≤ 35% for a tight precision control.
Results and Discussion
Optimization of ddPCR reaction
An established qPCR method can, at least partially, be transferred directly to ddPCR format, including the determined probe and primer designs. The PCR reaction parameters, however, still require optimization to achieve the best reaction efficiency now in a much smaller compartment compared with the bulk PCR volume.
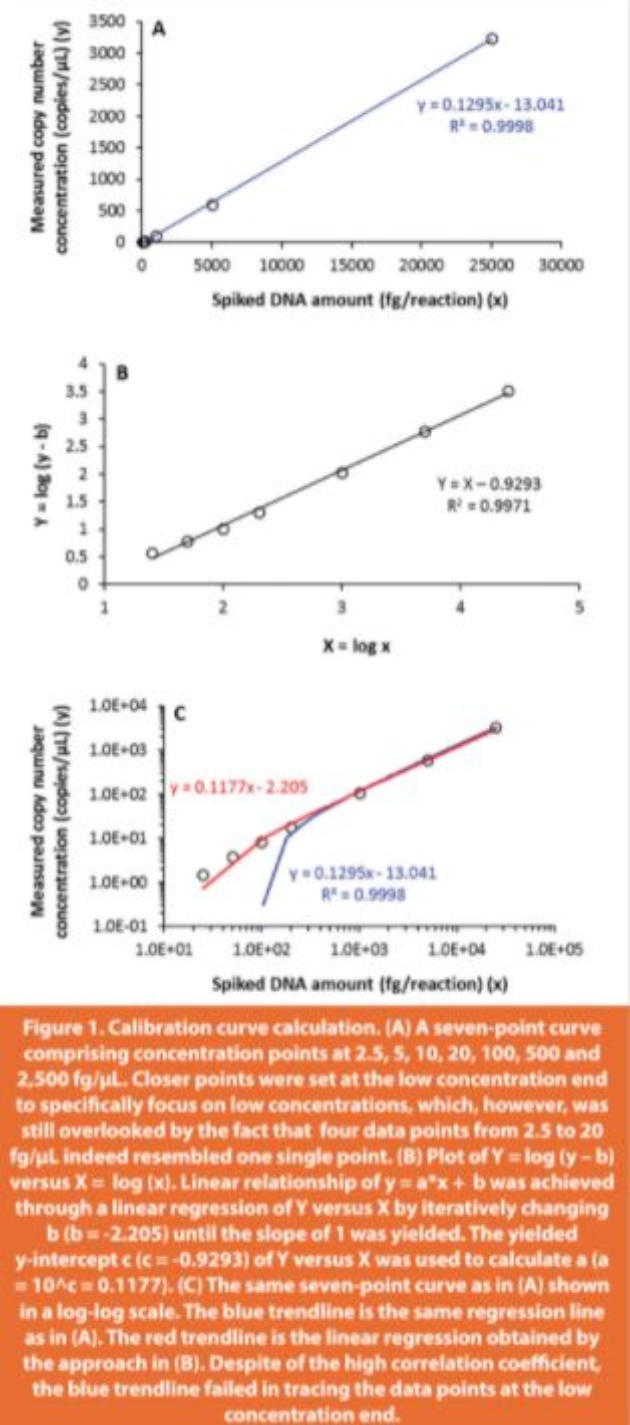
The annealing temperature of the PCR reaction is one of the most critical parameters for reaction efficiency and specificity.40 In ddPCR, the optimal annealing temperature provides the largest fluorescence amplitude difference between the positive and negative droplets, and minimizes the influence from nonspecific amplifications. To determine the optimal annealing temperature for the ddPCR reaction of salmon sperm DNA, a range of temperatures from 51.4 to 64.4°C were tested that were below and above the annealing temperature (60°C) used in qPCR assay15 (Figure 2). At high temperatures (64.4 and 63.6°C), the positive and negative droplets were poorly separated. Starting at 61.9°C, better separation was observed visually, but a non-specific amplification population shown by the extra cluster right above the negative droplets was also noted. As the temperature decreased, the extra cluster gradually merged with the positive droplets. Accordingly, 59.4°C was selected as the optimal annealing temperature that achieved the largest amplitude difference between the positive and negative droplets and the closest distance between the non-specific cluster and the negative droplets. Thus, the undesired clusters were excluded from the target quantification by setting the threshold above the cluster.
Pretreatment of ProS
DNA extraction is frequently performed in standard PCR approaches to remove matrix interferences prior to PCR reaction. High efficiency extraction of trace amounts of resDNA from a drug formulation with high-levels of active pharmaceutical ingredient and excipients can be time-consuming and inefficient extraction can lead to an inaccurate assessment of the true amount of resDNA in the samples. One of the hallmarks of ddPCR is enhanced tolerance to interfering substances achieved by a massive sample partitioning that dilutes background and non-target DNA molecules present in individual droplets. The direct ddPCR method has been successfully applied to resDNA quantification in a few protein drugs without the need for DNA extraction where the drug was added to the ddPCR reaction either directly34, 35 or after protein digestion.36
For this work, ProS was tested to see if ProS could be added directly to the ddPCR reaction of salmon sperm DNA without creating interference. Four samples were analyzed in a single experiment including no-template control (NTC), salmon sperm DNA (DNA), DNA plus 3 μg of ProS (DNA + 3 μg ProS), and DNA plus 0.3 μg of ProS (DNA + 0.3 μg ProS) per reaction (Figure 3). In the DNA alone sample, the positive and negative droplets were well separated with a good fluorescence signal for positive droplets and low fluorescence for negative droplets (Figure 3A). When 3 μg of ProS was added, the fluorescence signal largely diminished concordant with a drop in the number of droplets generated (Figure 3B). When a smaller amount (0.3 μg) of ProS was added, although the droplet generation was less significantly impacted, the fluorescence signal of positive droplets remained low, resulting in their merging with negative droplets. Further reductions in ProS amounts that reduced assay sensitivity (presented as resDNA per mg or per dose of ProS) were not tested.

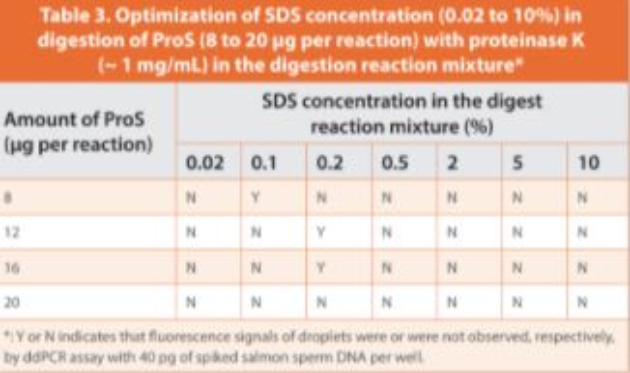
The matrix interference of ProS was attributed to the strong DNA binding capacity of the polycationic peptides. As protease digestion could release DNA from binding by disrupting the interaction between cationic arginine residues and DNA molecules, digested ProS was added to the reaction. The serine protease trypsin was tested initially to digest ProS because the trypsin catalytic triad contains a negative aspartate residue attracting positively charged lysine and arginine for cleavage, and is an endopeptidase that cleaves within the polypeptide chain.41 As expected, a better fluorescence signal was observed in some trypsin digested ProS samples in comparison to untreated ProS (unpublished data), however the digestion lacked reproducibility. As an alternative, proteinase K, one of the most active serine proteases with a full enzymatic activity in a broad pH range (from 6.5 up to 12) was tested. Since proteinase K can be stimulated by addition of denaturing agents such as sodium dodecyl sulfate (SDS),22 we examined the ratio of ProS to SDS by varying the concentration of SDS (0.02 to 10%) and the ProS load (8 to 20 μg per reaction) in the digestion reaction mixtures. Out of 28 combinations, three significantly reduced ProS interference (Table 3), demonstrated by high fluorescence of positive droplets and good separation of positive and negative droplets. Accordingly, the optimal SDS concentration in the digestion reaction mixture was set at 0.2%, and the maximum ProS load per reaction was set to 15 μg (which was at least 50-fold higher than the tolerable load of ProS (< 0.3 μg) when untreated).
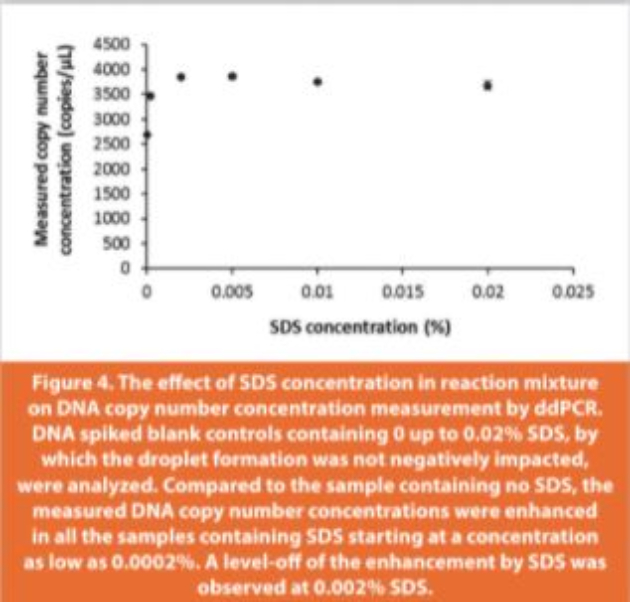
The recovery of spiked DNA from sample matrix containing digested ProS was tested by spiking salmon sperm DNA into either ProS (15 μg/ reaction) or TE buffer (blank control, zero load of ProS) in the presence of proteinase K and 0.2% SDS. Surprisingly, droplets failed to generate in the blank control (ProS free). In this case SDS, as an ionic surfactant, might impact the water-oil emulsion from which the water-in-oil droplets are formed, which would be counteracted by the presence of positively charged ProS. When the SDS concentration was reduced (0 to 0.02%), droplets formed normally without any suppression by SDS in blank controls. However, an unexpected enhancement was observed on the measured DNA copy number concentration in the presence of a small amount of SDS (0.001% to 0.02%) in comparison to the absence of SDS in the ddPCR reaction (Figure 4). Given this enhancement induced by SDS, the use of a blank control containing no SDS would likely overestimate the DNA recovery from ProS digested in the presence of SDS. Therefore, 0.02% SDS was selected for the blank control because 0.02% was the highest amount that did not impact the droplet formation, and the enhancement by SDS plateaued between 0.002% and 0.02%.
The necessity of adding proteinase K (present in ProS digestion reaction mixture) or saline (present in formulation of ProS drug product) to the selected blank control containing 0.02% SDS was examined. Neither the presence (or absence) of proteinase K nor saline altered the observed DNA copy number concentration among the three tested blank controls (Figure 5). TE buff er containing 0.02% SDS only was therefore used as the blank control in the assay (Table 2).
Evaluation of assay LLOQ, accuracy and dynamic range
ddPCR measures absolute DNA quantities by counting DNA-encapsulated droplets that are assigned by a threshold of fluorescence signal as positive droplets (containing one or more copies of intended DNA template) or negative droplets (containing no DNA template). The droplet counts are used to calculate the concentration of the target DNA in the sample using Poisson statistics. A calibration curve of DNA standard is, therefore, not required for the absolute quantification of DNA by ddPCR. However, for ddPCR, a DNA standard spike-in test using known quantities of DNA standard to identify the assay accuracy, precision, LLOQ and dynamic range is recommended.40 In addition, a calibration curve (i.e., measured DNA copies vs. known DNA amounts spiked) can be used to convert the copy number concentration (in copies/μL) measured by ddPCR to the weight (in fg) of DNA. Once the calibration curve is obtained, the measured copies of DNA in an unknown sample could be simply converted to the weight of DNA without any DNA standard in the experimental run.

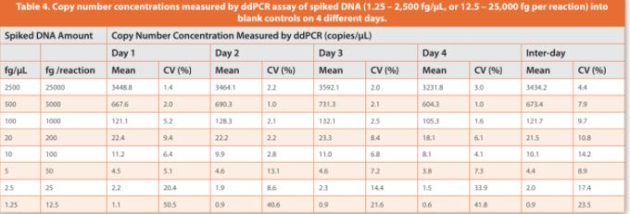
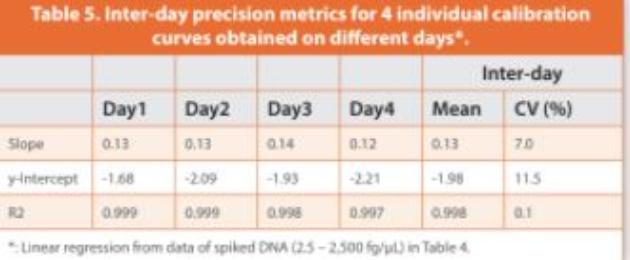
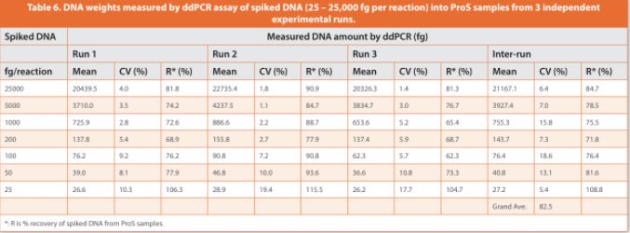
An initial eight-point DNA concentration curve (1.25 to 2,500 fg/ μL, or 12.5 to 25,000 fg of DNA per reaction) was used to establish the assay precision, LLOQ, linearity and dynamic range with four technical replicates per concentration, which was repeated four times on diff erent days. Intra-day coeffi cient of variation (CV) > 50% was observed at the lowest DNA concentration (1.25 fg/μL, or 12.5 fg per reaction), whereas intra-day CVs < 35% and inter-day CVs < 20% were observed for DNA concentrations at 2.5 fg/μL and above (Table 4). Accordingly, the lowest 1.25 fg/μL concentration point was excluded, and the DNA concentration range from 2.5 to 2,500 fg/μL was used to establish a seven-point calibration curve, with relatively closer points set for low concentrations where the resDNA amounts most likely reside. The inter-day CVs of the slope and y-intercept of linear regressions were within 15% across the four independent calibration curves established at diff erent days (Table 5). Collectively, the data showed a good linearity (R2 > 0.998), a LLOQ of 2.5 fg/μL (i.e., 25 fg per reaction, Table 1), and a three-order magnitude dynamic range (2.5 to 2,500 fg/μL, or 25 to 25,000 fg per reaction) for the salmon sperm DNA calibration curves by ddPCR assay.
The assay accuracy was determined by % recovery of spiked-in DNA from ProS sample matrices across the entire range of spiked amounts (25 to 25,000 fg per reaction). Digested spike-in ProS samples (15 μg of ProS per reaction) had good fl uorescence signals and eff ective separation between positive and negative droplets (Figure 6), indicating the successful removal of any matrix interference induced by untreated ProS (as shown in Figure 3). Using the established calibration curve from Table 5 (DNA copy number concentration (copies/μL) = DNA amount per reaction (fg) * 0.13 – 1.98), the measured DNA copies in spike-in ProS samples were converted to DNA weights in fg to evaluate the % recovery of DNA. The assay precision was demonstrated by CVs of the determined DNA weights within 20% for both intra-runs (four replicates per concentration) and inter-runs (three independent runs at diff erent days) across all the tested DNA amounts spanning three orders of magnitude starting from the lowest 25 fg per reaction (Table 6). Moreover, the % recovery of DNA from spike-in ProS samples was > 60% for intra-runs and 70 – 110% for inter-runs. There was also a high correlation between the measured DNA weights and the spiked-in DNA amounts in ProS samples (R2 =0.9998), which was closely related to the linear trendline for blank controls (ProS-free), indicating that the assay was largely free of matrix interference from the presence of ProS (Figure 7). In summary, these results demonstrate a validated assay LLOQ at 25 fg of DNA per reaction in the presence of 15 μg of ProS that is equivalent to 1.67 pg DNA/mg ProS (ppb) or 83.3 pg DNA per 50 mg dose of ProS (Table 1).
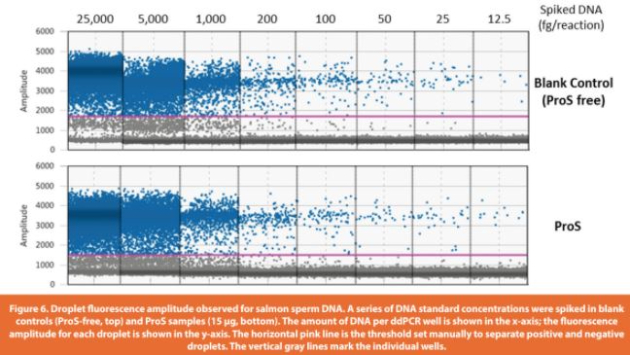

3.4 Measurement of resDNA in ProS drug products
Five lots of ProS for injection (10 mg/mL) purchased from the US market were analyzed by the developed ddPCR assay. Four vials from four different lots, and four vials from the same lot were analyzed. For all eight ProS samples, a near-zero copy number reading was obtained for each individual replicate, which was far below the copy numbers obtained at LLOQ (25 fg of DNA per reaction) (Figure 8A). This result shows that resDNA contamination in the tested ProS lots was well below the assay LLOQ.


A spike-in test (200 fg of DNA per reaction) was also performed on the 8 ProS samples. Compared to the ProS-free blank controls, the copy number concentrations measured for the ProS samples were on average 77.7% ± 6.7% and 76.2% ± 9.0% for intra-lots and inter-lots, respectively (Figure 8B). The small intra-lot or inter-lot variances indicate an undifferentiable matrix effect from lot-to-lot or vial-to-vial. This result is also in agreement with the % recovery within 70 – 110% with an overall average of 82.5% for spike-in ProS samples across the entire range of spiked DNA amounts (Table 6).
Conclusions
The low acceptance levels of resDNA in biopharmaceutical products, together with the high doses and complex matrixes, impose challenges for accurately quantifying resDNA contamination, especially in drugs like ProS with DNA binding avidity. In this study, a ddPCR method was adapted to quantify resDNA in ProS drug products by transferring a previously established qPCR assay into ddPCR format with modifications. The developed ddPCR assay detects resDNA at levels as low as 1.67 pg/mg (ppb) of ProS or 83 pg per 50 mg dose (LLOQ) with an average % recovery of 82.5% and a wide dynamic range, without DNA extraction. This LLOQ is 120 fold more sensitive than the acceptable resDNA amount (10 ng per dose), and is also below the guideline (100 pg per dose8) for particular products where cellular DNA is considered to pose a great risk (e.g., retroviral proviral sequences). This type of method can be applicable in resDNA testing at different stages of manufacturing (e.g., process intermediates, drug substance or drug product). Five lots of ProS drug products were analyzed using this method and none tested positive for resDNA contamination above the method’s LLOQ. Furthermore, this ddPCR format utilizes 96-well PCR plates throughout the procedure (from sample pretreatment to PCR assay), and can therefore be potentially complemented into an automated analytical platform by accommodating a robotic sample handling system for high-throughput and robust resDNA analysis in this drug class or other therapeutic protein drugs (where the protease digestion step prior to ddPCR reaction may or may not be needed).
Funding sources
Food and Drug Administration Regulatory Science and Review Enhancement (RSR) program provided partial funding for the study (to K.Y.).
References
- Oo, C.; Kalbag, S. S., Leveraging the attributes of biologics and small molecules, and releasing the bottlenecks: a new wave of revolution in drug development. Expert Rev Clin Pharmacol 2016, 9 (6), 747-9.
- Chan, J., & Chan, A., Biologics and biosimilars: what, why and how? ESMO Open 2017, 2 (1), e000180. doi:10.1136/esmoopen-2017-000180.
- Kesik-Brodacka, M., Progress in biopharmaceutical development. Biotechnol Appl Biochem 2018, 65 (3), 306-322.
- Rathore, A. S.; Sobacke, S. E.; Kocot, T. J.; Morgan, D. R.; Dufi eld, R. L.; Mozier, N. M., Analysis for residual host cell proteins and DNA in process streams of a recombinant protein product expressed in Escherichia coli cells. J Pharm Biomed Anal 2003, 32 (6), 1199-211.
- Wang, W.; Ignatius, A. A.; Thakkar, S. V., Impact of residual impurities and contaminants on protein stability. Journal of pharmaceutical sciences 2014, 103 (5), 1315-30.
- Yang, H.; Zhang, L.; Galinski, M., A probabilistic model for risk assessment of residual host cell DNA in biological products. Vaccine 2010, 28 (19), 3308-11.
- Wang, X.; Morgan, D. M.; Wang, G.; Mozier, N. M., Residual DNA analysis in biologics development: review of measurement and quantitation technologies and future directions. Biotechnol Bioeng 2012, 109 (2), 307-17.
- Yang, H., Establishing acceptable limits of residual DNA. PDA J Pharm Sci Technol 2013, 67 (2), 155-63.
- Rochon, A. G.; Belisle, S.; Couture, P.; Fortier, A.; Lebon, J. S.; Deschamps, A., In Vivo Protamine Titration Using Activated Coagulation Time to Neutralize HeparinAnticoagulation in Cardiac Surgery: Proof of Concept. J Cardiothorac Vasc Anesth 2020.
- Mahan, C. E., A 1-year drug utilization evaluation of protamine in hospitalized patients to identify possible future roles of heparin and low molecular weight heparin reversal agents. J Thromb Thrombolysis 2014, 37 (3), 271-8.
- Hecht, P.; Besser, M.; Falter, F., Are We Able to Dose Protamine Accurately Yet? A Review of the Protamine Conundrum. J Extra Corpor Technol 2020, 52 (1), 63-70.
- Boer, C.; Meesters, M. I.; Veerhoek, D.; Vonk, A. B. A., Anticoagulant and side-effects of protamine in cardiac surgery: a narrative review. Br J Anaesth 2018, 120 (5), 914-927.
- Applefield, D.; Krishnan, S., Protamine. In StatPearls, StatPearls Publishing LLC.: Treasure Island (FL), 2020.
- Solovyev, A. Y.; Tarnovskaya, S. I.; Chernova, I. A.; Shataeva, L. K.; Skorik, Y. A., The interaction of amino acids, peptides, and proteins with DNA. Int J Biol Macromol 2015,78, 39-45.
- Sommers, C.; Rosenzweig, B.; Oum, L.; Thompson, K.; Keire, D. A., Quantitation of residual host cell DNA in protaminesulfate drug product by qPCR. J Pharm Biomed Anal 2018, 160, 238-243.
- Oskolkov, N.; Bar-Shir, A.; Chan, K. W.; Song, X.; van Zijl, P. C.; Bulte, J. W.; Gilad, A. A.; McMahon, M. T., Biophysical Characterization of Human Protamine-1 as a Responsive CEST MR Contrast Agent. ACS Macro Lett 2015, 4 (1), 34-38.
- Roque, A.; Ponte, I.; Suau, P., Secondary structure of protamine in sperm nuclei: an infrared spectroscopy study. BMC Struct Biol 2011, 11, 14.
- Awotwe-Otoo, D.; Agarabi, C.; Keire, D.; Lee, S.; Raw, A.; Yu, L.; Habib, M. J.; Khan, M. A.; Shah, R. B., Physicochemical characterization of complex drug substances: evaluation of structural similarities and differences of protamine sulfate from various sources. AAPS J 2012, 14 (3), 619-26.
- Chang, J. T.; Chen, Y. C.; Chou, Y. C.; Wang, S. R., Quantitative detection of residual porcine host cell DNA by real-time PCR. Biologicals 2014, 42 (2), 74-8.
- Hu, B.; Sellers, J.; Kupec, J.; Ngo, W.; Fenton, S.; Yang, T. Y.; Grebanier, A., Optimization and validation of DNA extraction and real-time PCR assay for the quantitative measurement of residual host cell DNA in biopharmaceutical products. J Pharm Biomed Anal 2014, 88, 92-5.
- Hussain, M., A direct qPCR method for residual DNA quantification in monoclonal antibody drugs produced in CHO cells. J Pharm Biomed Anal 2015, 115, 603-6.
- Peper, G.; Fankhauser, A.; Merlin, T.; Roscic, A.; Hofmann, M.; Obrdlik, P., Direct real-time quantitative PCR for measurement of host-cell residual DNA in therapeutic proteins. J Pharm Biomed Anal 2014, 100, 123-130.
- Zhang, W.; Wu, M.; Menesale, E.; Lu, T.; Magliola, A.; Bergelson, S., Development and qualification of a high sensitivity, high throughput Q-PCR assay for quantitation of residual host cell DNA in purification process intermediate and drug substance samples. J Pharm Biomed Anal 2014, 100, 145-149.
- Sommers, C.; Rosenzweig, B.; Oum, L.; Thompson, K.; Keire, D. A., Development of methods for data quantitation of spiked salmon host cell DNA in protamine sulfate by qPCR. Data Brief 2018, 21, 644-652.
- Garibyan, L.; Avashia, N., Polymerase chain reaction. J Invest Dermatol 2013, 133 (3), 1-4.
- Navarro, E.; Serrano-Heras, G.; Castano, M. J.; Solera, J., Real-time PCR detection chemistry. Clin Chim Acta 2015, 439, 231-50.
- Cave, L.; Brothier, E.; Abrouk, D.; Bouda, P. S.; Hien, E.; Nazaret, S., Efficiency and sensitivity of the digital droplet PCR for the quantification of antibiotic resistance genes in soils and organic residues. Appl Microbiol Biotechnol 2016, 100 (24), 10597-10608.
- Hindson, B. J.; Ness, K. D.; Masquelier, D. A.; Belgrader, P.; Heredia, N. J.; Makarewicz, A. J.; Bright, I. J.; Lucero, M. Y.; Hiddessen, A. L.; Legler, T. C.; Kitano, T. K.; Hodel, M. R.; Petersen, J. F.; Wyatt, P. W.; Steenblock, E. R.; Shah, P. H.; Bousse, L. J.; Troup, C. B.; Mellen, J. C.; Wittmann, D. K.; Erndt, N. G.; Cauley, T. H.; Koehler, R. T.; So, A. P.; Dube, S.; Rose, K. A.; Montesclaros, L.; Wang, S.; Stumbo, D. P.; Hodges, S. P.; Romine, S.; Milanovich, F. P.; White, H. E.; Regan, J. F.; Karlin-Neumann, G. A.; Hindson, C. M.; Saxonov, S.; Colston, B. W., High- throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011, 83 (22), 8604-10.
- Pinheiro, L. B.; Coleman, V. A.; Hindson, C. M.; Herrmann, J.; Hindson, B. J.; Bhat, S.; Emslie, K. R., Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal Chem 2012, 84 (2), 1003-11.
- Taylor, S. C.; Laperriere, G.; Germain, H., Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: from variable nonsense to publication quality data. Sci Rep 2017, 7 (1), 2409.
- Hayden, R. T.; Gu, Z.; Ingersoll, J.; Abdul-Ali, D.; Shi, L.; Pounds, S.; Caliendo, A. M., Comparison of droplet digital PCR to real-time PCR for quantitative detection of cytomegalovirus. J Clin Microbiol 2013, 51 (2), 540-6.
- Campomenosi, P.; Gini, E.; Noonan, D. M.; Poli, A.; D’Antona, P.; Rotolo, N.; Dominioni, L.; Imperatori, A., A comparison between quantitative PCR and droplet digital PCR technologies for circulating microRNA quantification in human lung cancer. BMC Biotechnol 2016, 16 (1), 60.
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z., Comparison of Droplet Digital PCR and Quantitative PCR Assays for Quantitative Detection of Xanthomonas citri Subsp. citri. PLoS One 2016, 11 (7), e0159004.
- Anderson, J. a. H., M., A Direct Droplet Digital PCR Method for E. coli Host Residual DNA Quantification. Pharmacology & Pharmacy 2018, 9, 117-123.
- Hussain, M.; Fantuzzo, R.; Mercorelli, S.; Cullen, C., A direct droplet digital PCR method for quantification of residual DNA in protein drugs produced in yeast cells. J Pharm Biomed Anal 2016, 123, 128-31.
- Hussain, M. B., J., A Droplet Digital PCR Method for CHO Host Residual DNA Quantification in Biologic Drugs. J Anal Pharm Res 2017, 4 (3), 00107. DOI: 10.15406/japlr.2017.04.00107.
- Wang, Y.; Cooper, R.; Bergelson, S.; Feschenko, M., Quantification of residual BHK DNA by a novel droplet digital PCR technology. J Pharm Biomed Anal 2018, 159, 477-482.
- Jiang, X.; Yang, K.; Han, X., Direct quantitation of psychosine from alkaline-treated lipid extracts with a semi-synthetic internal standard. J Lipid Res 2009, 50 (1), 162-72.
- USP <1130> Nucleic Acid-based Techniques - Approaches for Detecting Trace Nucleic Acids (Residual DNA Testing). USP-NF 2016.
- Valpione, S.; Campana, L., Detection of circulating tumor DNA (ctDNA) by digital droplet polymerase chain reaction (dd-PCR) in liquid biopsies. Methods Enzymol 2019, 629, 1-15.
- Teaster T. Baird, J. a. C. S. C., Trypsin. In Handbook of Proteolytic Enzymes, 3rd ed.; Neil D. Rawlings, G. S., Ed. Elsevier Ltd.: 2013; pp 2594-2600.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox! Sign up now!