Pete Clarner1*, Katelyn M. Mansfield2*
1 Gene Therapy Accelerator Unit, Biotherapeutics and Medicinal Sciences, Biogen Inc.
2 Analytical Development, Product and Technology Development, Biogen Inc.
*Both authors contributed equally
Abstract
Gene therapy is a modality that requires a complex analytical control strategy to ensure safety and efficacy of the drug product. Adeno-associated virus (AAV) is widely used as a vector for preclinical research and clinical gene therapy development. AAV gene therapies require characterization of many product quality attributes such as the quantitation of genome titer and aspects of AAV potency such as internalization, infectivity, expression, and functional potency (Figure 1). PCR is one of the most well-known and widely used methods to quantitatively analyze AAV gene therapy candidates at all stages of development. Here, we discuss the many uses of PCR-based technologies as an analytical tool for characterizing AAV gene therapies.
Detection Technologies
The two key detection technologies used for PCR-based quantitative methods are SYBR and TaqMan (Figure 2). Each requires the development of a sequence-specific forward and reverse primer. SYBR is a chelating dye that significantly increases in fluorescence when bound to double-stranded DNA (dsDNA), including newly formed PCR product dsDNA.1
TaqMan technology uses a third DNA oligo, the sequence-specific hydrolysis probe. The probe has a fluorescent reporter dye on the 5’ end and a quencher on the 3’ end. When the probe is bound to the target sequence, 5’ exonuclease activity of the DNA polymerase cleaves the reporter dye from the probe, resulting in separation from the quencher and fluorescent signal. Several technologies are available to improve primer/probe performance such as the addition of a minor groove binder (MGB) to increase melting temperature, enabling shorter, more target-specific probes, or an additional internal quencher to reduce background signal.
While SYBR is lower in cost, TaqMan has a higher level of specificity and sensitivity, and allows for duplexing or multiplexing. SYBR binds non-specifically to all dsDNA, so false positive signals are possible via primer dimers or other non-target DNA.1,2
qPCR Versus dPCR
The two main platforms utilized for PCR-based quantitative methods are quantitative real-time PCR (qPCR) and digital PCR (dPCR). In qPCR each reaction well is a single fluorescent signal that is proportional to the amount of starting target DNA present in the reaction mixture. On the other hand, dPCR employs the separation of the reaction mixture and sample into thousands of partitions. Specifically, in droplet digital PCR (ddPCR), these partitions are water-in-oil droplets. Based on a fluorescence amplitude threshold, droplets can be scored as positive or negative for the PCR reaction occurring. Using a Poisson distribution calculation, the proportion of positive and negative droplets can then be used to determine absolute concentration of the target DNA sequence.
Although qPCR has the advantage of being a less expensive PCR method and it can be miniaturized to 384-well, ddPCR has several advantages to qPCR including that it does not require a standard curve, is less influenced by PCR bias because of matrix interference or other PCR inefficiencies, and has a high level of sensitivity and precision. Therefore, ddPCR is the preferred method for absolute quantitation of in-process intermediates that may have different buffer and matrix compositions.3 It also has become the platform of choice for many applications in gene therapy.3,4,5
One-Step Versus Two-Step Methods for RNA Detection
RNA levels can be measured by isolating total RNA and reverse transcribing it to cDNA. The resulting cDNA is quantified via PCR. Depending on the type of target to be measured, different reverse transcription (RT) and PCR protocols can be used. RT-qPCR and RT-ddPCR methods may utilize either a one-step method, whereby the reverse transcription and subsequent qPCR or ddPCR occur in the same reaction mix, or two-step where the cDNA is generated in a reverse transcription reaction and then subsequently quantified by qPCR or ddPCR. One-step PCR methods typically use the reverse primer from the detection primer/probe set for the RT step. This method reduces the number of steps required to quantitate RNA expression and involves less sample handling and pipetting steps, thus reducing assay variability and possibilities for sample contamination. One caveat is that one-step methods do not generate a stock of cDNA for later use, preventing potential for re-analysis. One-step methods also limit the ability to separately optimize the RT and PCR reactions. Although two-step PCR methods require an additional step, they allow for a stock of cDNA to be generated for multiple PCR reactions. This can be useful for method troubleshooting as well as allowing both steps to be fully optimized independently. Two-step methods can employ several different types of primer for the RT step including the reverse primer from the detection primer/probe set, oligo(dT) primers, or random primers.
For qPCR methods, with low starting material or low abundant targets, two-step methods can provide better sensitivity and more accurate quantitation due to the ability to titrate cDNA levels for the PCR reaction. This allows for diluting PCR inhibitors that may be carried over from the RNA isolation and RT reaction. However, for ddPCR one-step and two-step methods are less prone to influence by matrix effects and have similar sensitivity.4
Characterization of AAV Using PCR Technologies
AAV gene therapy analytical control strategies necessitate characterization of the AAV at multiple stages throughout the transduction pathway. Below we discuss how PCR-technologies can be utilized for many aspects of this characterization for both in vitro and in vivo studies(Figure 1).
AAV Genome Titer
One of the most important analyses performed for AAV vectors is determining the physical quantity as represented by genome titer. AAV genome titer is used to define dosing in preclinical animal models as well as in patients in the clinic. Additionally, genome titer is critical to support efforts of process development groups, to release batch material, and to perform other analytical assays, such as potency assays. Both qPCR and ddPCR platforms have been applied to determining AAV genome titer.3,5,6 qPCR has several disadvantages for this application including the requirement of a well calibrated standard curve and the influence of PCR bias on quantitative results. Standard curves for qPCR-based methods could be linearized plasmid containing the target sequence or a reference standard AAV with a defined genome titer.6 One of the additional challenges is that qPCR assays can have a wide lab-to-lab variability.7 The dependence of a qPCR-based method on a standard can also be challenging if that standard is changed throughout the lifecycle of the method or a program; particularly if the method has been used for dosing in clinical trials and the change of standard results in different quantitation such as shown in the example in Table 1.
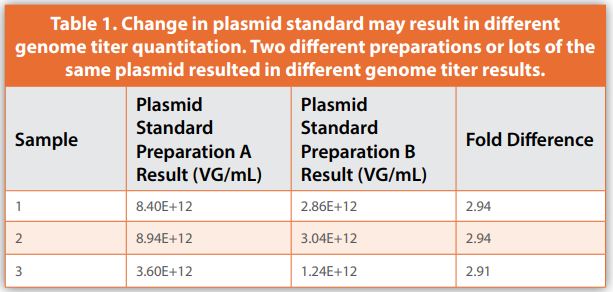
How the standard is quantified for its value assignment can also impact the quantitative results it generates. For example, Table 2 shows a linearized plasmid standard quantified in ng/µL by two common dsDNA detection methods, PicoGreen assay and UV absorbance by NanoDrop. Each method generates different values, which when converted to a copies per volume value, have a significant difference.
Because of the challenges associated with qPCR methods, ddPCR-based assays are being more widely adopted. They have proved to be highly precise and provide absolute quantitation, rather than relying on a standard curve .3.5
Infectivity
A platform approach to determining one attribute of AAV vector activity is to measure its infectious titer or infectivity. This type of method can be applied to virtually any AAV vector. The most common method for determining AAV infectivity is a 50% tissue culture infectious dose (TCID50) assay. TCID50 utilizes a stable cell line, HeLaRC32, which expresses AAV2 rep and cap genes. These cells are co-infected with AAV and adenovirus type 5 (Ad5), resulting in the replication of infectious AAV particles within the cells. Replicated AAV can then be detected by PCR and infectious titer is calculated using the Spearman-Kärber method. Utilizing endpoint dilutions, single infection events can be detected using the TCID50 method.8 The generated result is an absolute value represented as TCD50/mL or infectious units per mL (IU/mL). While the assay can show biological activity of AAV particles, it has several drawbacks. Different AAV capsids will have varying tropism to the HeLaRC32 cells used in the assay system and the absolute value result may not truly be representative of how infectious an AAV vector is. Additionally, the absolute value result can be quite variable. This leads to reproducibility challenges within the same lab and between labs, making assay transfer difficult.7 The replicative ability of the AAV vector in the assay system is also not reflective of what occurs when the vector is administered in vivo, as recombinant AAV vectors for gene therapy are replication incompetent. Therefore, assays that measure cellular uptake or internalization via PCR may be more relevant for AAV therapies. Additionally, relative internalization assays which measure the AAV transgene relative to a housekeeping gene would have a higher level of precision and robustness as opposed to an absolute measurement assay. Furthermore, the TCID50 assay does not demonstrate vector-specific potency either at the expression or functional level. Program-specific assays are required for AAV products as they advance into clinical trials and this necessitates development of additional functional assays to support AAV gene therapy programs.
PCR for Expression and Potency (RT-PCR, RT-ddPCR)
Potency of a gene therapy product is dependent, in part, on how well the transgene is expressed. Therefore, measuring transgene expression levels in transduced cells is an important analytical method for all gene therapy products. PCR-based methods can be used to detect transgene RNA expression levels for many types of gene therapies such as gene-augmentation and gene-silencing therapies. For gene-augmentation, the goal is to replace a missing or aberrant protein within the cell to modulate the disease state. The transgene typically contains a coding DNA sequence that gets transcribed into mRNA and translated into functional protein. In vitro expression of a gene-augmentation transgene is often quantified using RT-qPCR and/or RT-ddPCR for mRNA, or an immunoassay for protein.
In gene-silencing gene therapies, the transgene cassette expresses a small RNA, such as a miRNA or siRNA, to reduce the expression of an endogenous gene in the target cells to modulate the disease state. Analytical PCR methods may be used to quantitate expression levels of the small RNA expressed from the AAV genome, as well as measure the reduction of target mRNA levels. The most common PCR-based method for small RNA detection uses stem-loop primer reverse transcription followed by PCR quantitation.9
For gene-silencing strategies that involve knockdown of an endogenous gene target, quantitating the downregulation of target mRNA demonstrates the mechanism of action of the therapy and can demonstrate functional potency. This type of assay can either utilize PCR or immunoassays to quantitate the levels of the target mRNA or protein, respectively. Sometimes both assays may be developed during phase-appropriate development and may be used in parallel to show consistency between the orthogonal methods.
Process Related Analyses
PCR methods can be used to detect process-related impurities such as residual plasmid DNA, residual host cell DNA, and residual helper viruses. As these impurities may pose a risk to product quality and safety, their accurate detection and quantitation is required for all gene therapy products. These impurities arise during vector production and may remain in the product even after the purification process. The concentration of impurities in a product can be determined by PCR amplifying a sequence found in the impurity (e.g., antibiotic resistance sequence of a DNA plasmid). For process-related DNA impurities, the FDA recommends having less than 10ng of DNA per dose and that the size of the DNA is less than 200 base pairs to reduce potential oncogenicity and infectivity.10 In addition to quantifying the concentration of residual DNA in gene therapy products, 2-dimensional ddPCR (2D-ddPCR) can be used to determine the approximate fragment size by using primer sets to multiple areas along the sequence.
PCR is also a commonly used technique for assessing genome integrity of AAV. As AAV packaging may result in incomplete genomes, accurately measuring what has been packaged in the AAV is vital to ensuring product safety and efficacy. As previously stated, qPCR results can be affected by the standard curve material selected for the study, and therefore ddPCR is often preferred for genome integrity analyses as it results in absolute quantification of the target. 2D-ddPCR for genome integrity can quantify two separate parts of the same viral genome, therefore increasing the accuracy of detection of incomplete AAV genomes as compared to 1D-ddPCR.11
Additional Considerations
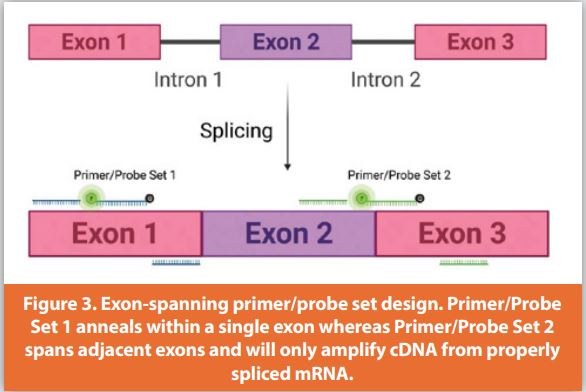
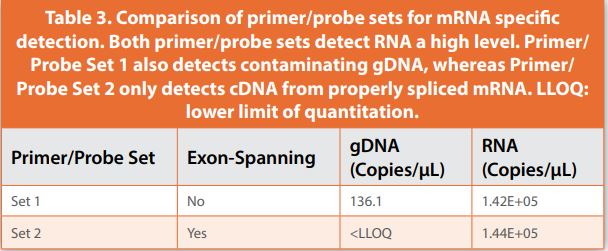
When measuring the expression level of endogenous mRNA, whether this is the reference gene or target gene within the cell, detection of contaminating genomic DNA (gDNA) from the cell can be mitigated through reagent design. By designing primer/probe sets that span two exons of the coding sequence of the endogenous gene, this will allow only quantitation of cDNA reverse transcribed from properly spliced mRNA (Figure 3). For example, two primer/probe sets, one annealing within a single exon and the other spanning adjacent exons, were compared for specificity, demonstrating that primer/probe sets within a single exon detected contaminating gDNA. In some cases, this can skew mRNA quantitation leading to less accurate expression results. (Figure 3, Table 3).
Depending on the intended application of a PCR method, enzymatic treatment of samples can be a crucial sample processing step. In genome titer applications, DNase digestion of non-encapsidated genomes and residual ITR plasmid are critical to ensure quantitation of only encapsidated vector genomes. For mRNA expression analysis of the AAV transgene either by RT-qPCR or RT-ddPCR, detection of contaminating DNA cannot be mitigated through exon-spanning primer/probe design. Therefore, efficient DNase removal of both viral ssDNA and dsDNA are essential to ensure only quantitation of cDNA reverse transcribed from transgene mRNA.
In vivo Applications
PCR methods can be utilized for characterizing AAV performance in vivo as well as in vitro. For biodistribution analyses, AAV vectors are dosed into preclinical animal models and following the in-life portion of the study, tissues are harvested and vector DNA can be analyzed with PCR technologies to assess vector delivery across different tissue types. Additionally, like in vitro analytical assays, tissues can be analyzed for vector mRNA expression as well as functional potency by quantifying target mRNA repression.4,12
Conclusion
PCR is a powerful technology for characterizing AAV molecules, but care must be taken in the assay design, execution, and interpretation of results to ensure assay accuracy and validity. This can be achieved through including proper controls and understanding the limitations and advantages of PCR technologies when characterizing AAVs. Selecting the appropriate platform and technology for a particular application is also an important part of the assay development process.
References
- Tajadini M, Panjehpour M, Javanmard SH. Comparison of SYBR Green and TaqMan methods in quantitative real-time polymerase chain reaction analysis of four adenosine receptor subtypes. Adv Biomed Res. 2014; 3:85. doi:10.4103/2277-9175.127998.
- Navarro E, Serrano-Heras G, Castaño MJ, Solera J. Real-time PCR detection chemistry. Clin Chim Acta. 2015;439:231-50. doi: 10.1016/j.cca.2014.10.017.
- Dobnik D, Kogovsek P, Jakomin T, et al. Accurate quantification and characterization of adeno-associated viral vectors. Front Microbiol. 2019;10:1570. doi: 10.3389/fmicb.2019.01570.
- Clarner P, Lau SK, Chowdhury T, et al. Development of a one-step RT-ddPCR method to determine the expression and potency of AAV vectors. Mol Ther Methods Clin Dev. 2021;23:68-77. doi: 10.1016/j.omtm.2021.05.003.
- Lock M, Alvira MR, Chen S, Wilson JM. Absolute determination of single-stranded and self-complementary adeno-associated viral vector genome titers by droplet digital PCR. Hum Gene Ther Methods. 2014;25(2):115-125. doi: 10.1089/hgtb.2013.131.
- Wang Y, Menon N, Shen S, Feschenko M, Bergelson S. A qPCR method for AAV genome titer with ddPCR-level of accuracy and precision. Mol Ther Methods Clin Dev. 2020;19:341-346. doi: 10.1016/j.omtm.2020.09.017.
- Lock M, McGorray S, Auricchio A, et al. Characterization of a recombinant adeno-associated virus type 2 reference standard material. Hum Gene Ther. 2010;21(10):1273-1285. doi: 10.1089/hum.2009.223.
- Zhen Z, Espinoza Y, Bleu T, Sommer JM, Wright JF. Infectious Titer Assay for Adeno-Associated Virus Vectors with Sensitivity Sufficient to Detect Single Infectious Events. Human Gene Therapy. 2004;15(7):709-715. doi: 10.1089/1043034041361262.
- Marcial-Quino J, Gomez-Manzo S, Fierro F, et al. Stem-loop RT-qPCR as an efficient tool for the detection and quantification of small RNAs in Giardia lamblia. Genes (Basel). 2016;7(12):131. doi: 10.3390/genes7120131.
- Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drug applications (INDs). FDA Guidance Documents. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigational-new-drug. Accessed December 20, 2021.
- Furuta-Hanawa B, Yamaguchi T, Uchida E. Two-dimensional droplet digital PCR as a tool for titration and integrity evaluation of recombinant adeno-associated viral vectors. Hum Gene Ther Methods. 2019;30(4):127-136. doi: 10.1089/hgtb.2019.031.
- De BP, Chen A, Salami CO, et al. In vivo potency assay for adeno-associated virus-based gene therapy vectors using AAVrh.10 as an example. Hum Gene Ther Methods. 2018;29(3):146-155. doi: 10.1089/hgtb.2017.246.
About the Authors
Pete Clarner, Scientist I, GTxAU, holds a B.S. in Biology from Boston College and a PSM in Biotechnology from Northeastern University. He joined Biogen in 2012 as a member of the Bioassay and Gene Therapy group of the Analytical Development department. In 2019 he joined the Gene Therapy Accelerator Unit (GTxAU) within R&D. Pete has extensive experience in AAV characterization and serves as the GTxAU SME, particularly in PCR-based methods development, with emphasis on the ddPCR platform.
Katelyn M. Mansfield, Scientist II, Technical Development, is a Scientist at Biogen. She received her PhD in Cell and Molecular Biology from Boston University and has been with the Analytical Development Department at Biogen since 2019. Kate specializes in the development of cell-based assays for Biogen’s biologics portfolio, including AAV gene therapies.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special offers
from American Pharmaceutical Review – all delivered right to your inbox!
Sign up now!