Li Li, Peter Tattersall, Qinggang Wang, Jonathan Shackman, Lianjia Ma and Adrian Doggett- Chemical Process Development, Bristol Myers Squibb
Overview
Active pharmaceutical ingredient (API) process development requires a comprehensive analytical control and a set of wide-ranged analytical methods. In the early to mid-stages of development, i.e., at phase I and II clinical trials, the analytical controls in manufacturing processes are still evolving. The analytical methods designed to gather data on APIs, intermediates, starting materials, and reagents are also progressing, but knowledge regarding method robustness is limited. Despite being in clinical development, there is a requirement for scientifically sound data to provide confidence in key decisions, establish reproducible testing at contract manufacturing organizations (CMOs), and, most importantly, guaranteeing consistent quality to ensure patient safety. As every project is different, how do we drive consistency between projects in handling and learning from risk mitigation? To address this, an analytical risk survey program to evaluate common risk factors was developed. In this article, we introduce a new workflow that is simple and widely applicable to assess analytical gaps and risks for early- to mid-stage projects. The workflow combines a checklist with common risk factors and a spreadsheet with scored ranking to facilitate the collation of an experimental ‘to do’ list for evolution of analytical method conditions and optimization of controls. The impact of this risk survey program has shown to be value added for both the analytical teams and overall portfolio development at Bristol Myers Squibb.
Introduction
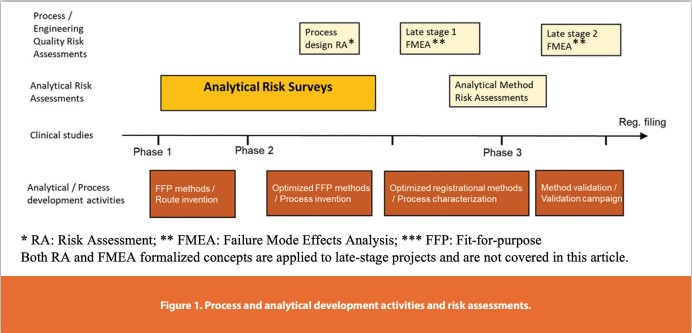
In the pharmaceutical industry, a scalable synthesis involves development of practical, safe, sustainable, and cost-effective chemistry and process design. The synthetic route, process, and analytical chemistry all evolve with the clinical development. At early clinical stages, i.e., phase I and II clinical trials, the process for small molecule APIs is usually grounded in the drug discovery route and fit for mid-scale (gram to 10’s of kilogram) production needs. At the late clinical stage, the chemical process should be well defined, robust for commercial regulatory filings, and ready for full commercial manufacturing scale. The analytical controls and methods of each stage follow these needs similarly. Analytical risk assessment should have the flexibility to meet the evolving demands of process development. To account for the analytical risk assessment needs, we designed a top-down approach which we call an “analytical risk survey”. This survey program is designed for early-stage projects, and the terminology of risk survey is utilized to differentiate a more involved risk assessment program for late-stage projects. In Figure 1 both process and analytical activities at different stages are summarized. Our analytical risk assessment program holistically views the analytical activities and how they collectively connect to the process to drive simple and effective method development. When viewed through the intent of the current analytical control strategy (not from an individual method), the risk survey helps highlight areas of concerns.
In general, the risk appraisal process contains hazard identification, risk analysis, and risk evaluation components. These concepts have been applied to many areas of business, government, and industry to identify any significant hazards to working projects.1 Our early development analytical risk survey process also utilizes these concepts. Risk and hazard identification is achieved by evaluating categories that include methods, knowledge gaps, control test placement, control test criteria, and impurity assessment as required by ICH guidelines Q3A, Q3C, Q3D and M7.2-5 The analysis methods that are used for characterizing organic impurities, inorganic impurities, residual solvents, and mutagenic impurities are evaluated with respect to being scientifically sound and suitable for their intended purpose. Some of these may be considered as critical quality attributes (CQAs). Organic impurity tests, including starting materials, by-products, intermediates, degradants, reagents, ligands, catalysts, enantiomers and mutagenic impurities are all considered to be and included in the common risk categories. Inorganic impurity tests, residual solvent analyses, in-process controls (IPCs), and manufacturing equipment cleaning methods may also be considered.
Once identified, risk analysis is the second important component in the risk appraisal process. Risk analysis is the assessment of the risks associated with possible hazards. It is the qualitative or quantitative process of evaluating the likelihood of occurrence and severity of harms.1 In our risk survey program, the method performance parameters and validation requirements in ICH Q2(R1)6 are used to evaluate the risk severity within each of the risk categories. A baseline performance of the analytical methods is reviewed to evaluate the severity of the risks. At early clinical stages, the control strategies are preferred to be objective and simple to execute, yet also aligned with ICH guidelines. Most analytical methods are fi t-for-purpose (FFP) at this stage, due to the evolving process chemistry. Therefore, our risk severity evaluation must consider these factors and make the risk analysis phase-appropriate.
The third component of risk assessment is risk evaluation. Here the evaluation attempts to define the estimated risk to analytical method performance. Assessment of the analytical methods can enable the introduction of controls that mitigate risk. Our risk evaluation is a combination of actual method performance and the stage-appropriate scientific and quality expectations. In our risk survey program, we compare the identified and analyzed risk characteristics against risk criteria previously collated based on cumulative experiences with historical projects. A subject matter expert (SME) committee reviews the risk estimation presented by the project team and together they adjust as needed. With the aligned risk, which is simply marked as low, medium, or high, the project team will work with SMEs to determine the actions required for risk mitigation.
Discussion
Risk acceptance of early-stage analytical methods
It is commonly understood that risk is defined as the combination of the probability of occurrence and the severity of a harm.1 Probability of analytical risk correlates strongly with the complexity of analytical methodologies (e.g., a gas chromatography method is more complicated than loss on drying) and also with an analyst’s performance or experience (i.e., more experience can generally reduce analytical risk.). Criteria for evaluating risk probability are defined in Table 1 rows with tan shading. Severity more closely correlates to the purpose of analytical tests (e.g., release control tests have higher risk than knowledge gathering tests), placement in a synthetic route (e.g., closer to a final API step is riskier than earlier steps), and stage of development or clinical trial (severity of risk is higher at later stages). Gauges to assess risk severity levels are defined in Table 1 rows with blue shading. These considerations or differentiators can be used as a tool for analytical teams to help assess their analytical risks (see Table 1).
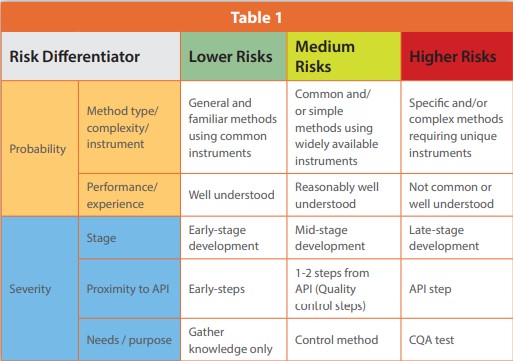
Figure 2 helps to visualize the “line of acceptable risk” generated using complexity of analytical methodology and performance risk with project stage.7 This highlights that risk tolerance at early stages is relatively larger and decreases as clinical development progresses, which Table 1 captures for risk survey assessors. The risk survey program is designed to meet the early stage needs by performing a “top down”, holistic analytical control evaluation. Late-stage projects, however, have much lower tolerance to analytical risks and must be suitable for routine quality control analysis, which is beyond the scope of this article.
Risk categories of early and mid-stage projects
Risk categories are utilized to aid in risk identification. The risk categories were collated from: molecular structure and related unique material properties; historical data, as would come from method transfer experience; ICH guideline requirements, including release specification guidance; and other drug substance analytical activities. Covered within these categories are:
- Method types, which can range from stability-indicating methods to specific trace impurity analysis (e.g., nitrosamines).
- Method analytical target profiles, which are typically linked to process control limits or release specifications.
- Physicochemical properties that can include presence of UV chromophores, solubility, hygroscopicity, static nature, etc.
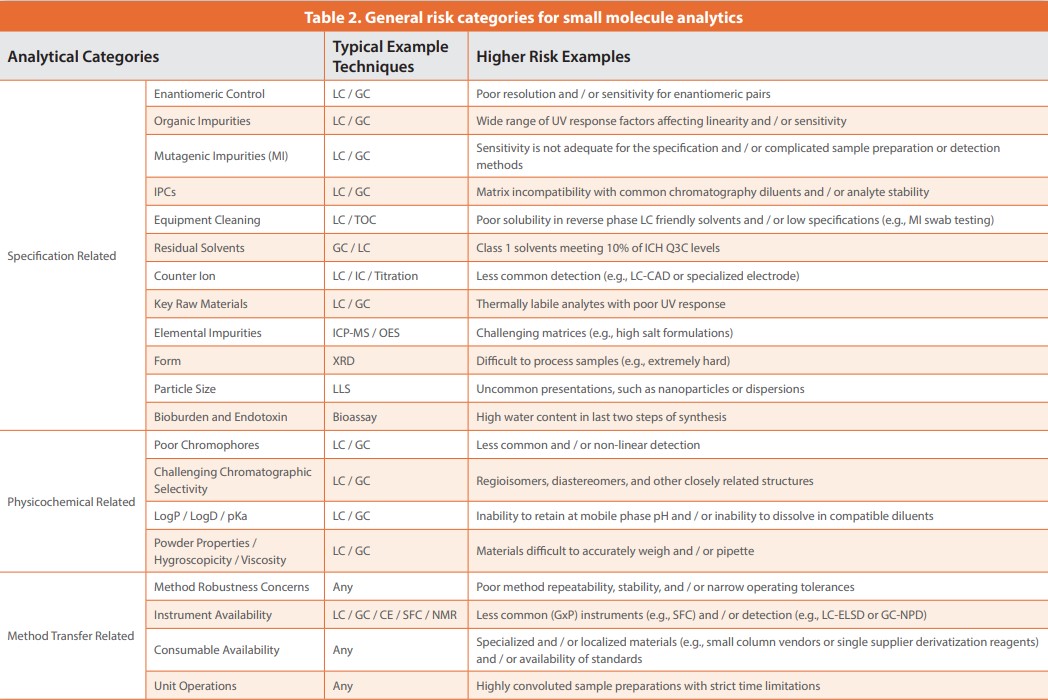
Table 2 lists the most common analytical risk categories we have encountered to help project teams evaluate soft spots in their controls and acts as a common check list to drive consistent practices across teams. Note, some of these categories may overlap, such as enantiomeric control and chromatographic selectivity.
Risk banding
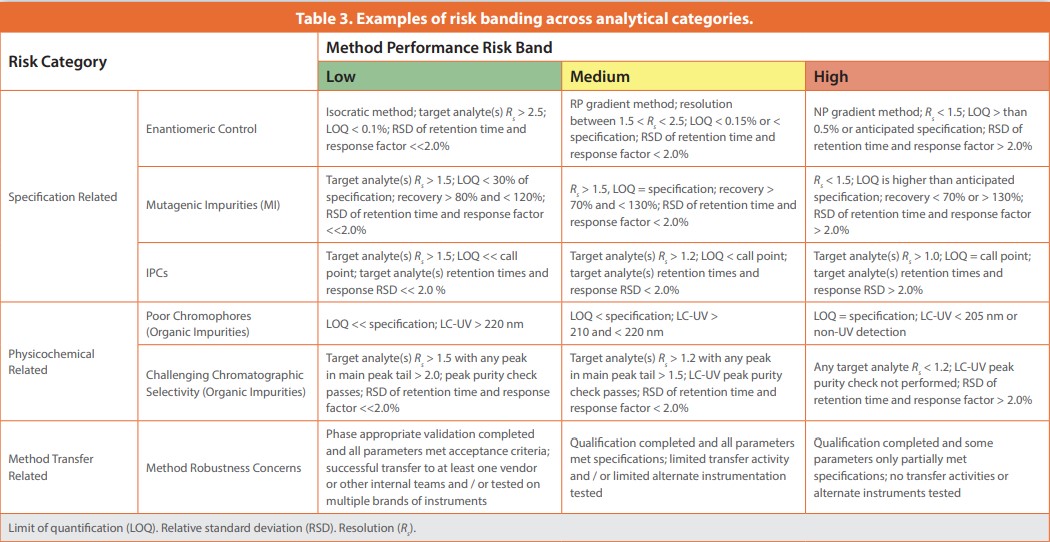
Risk analysis is the qualitative or quantitative process of measuring the likelihood of occurrence and severity of harms and must be performed across each of the identified risk categories. The evaluation of the harm and potential impact to quality should be based on scientific knowledge and ultimately link to the protection of the patient.1 In our analytical group, the risk level of each category is measured by comparing the analytical method’s observed performance to expected quality and scientific requirements, as well as historical experience. For example, widely spaced analytes in an isocratic LC method are much lower risk than a multiple stage gradient LC method just meeting baseline resolution requirements.
When the observed performance of the method is significantly better than the regulatory and scientific requirements, the risk level is banded as “low”. When the performance of the analytical method meets but approaches the limit of regulatory or scientific expectancy, the risk level is defined as “medium”. When the performance of analytical method fails to meet either regulatory or scientific expectancies, the risk level is defined as “high”. High risk scenarios can occur with rapidly changing projects and expectations. For example, process changes can generate entirely new impurity profiles, which may challenge existing methods. Even a defined process can become high risk if robustness studies performed near to the risk survey identify gaps (e.g, co-eluting impurities are found during a peak homogeneity check). Additionally, acceptable risk in the earliest phase may present as high risk if they are nearing a transfer or stage transition due to tightening requirements. Table 3 lists a few illustrative examples of risk banding. Over time, we have compiled the more common risk categories and criteria across method type combinations (e.g., LC-UV for organic impurities or GC-FID for residual solvents) to aid analytical teams’ assessments. The selected parameters are generally the more technically challenging aspects of the method or the more important quality aspects of the method. For less common category and method combinations, historical data is evaluated, literature is surveyed, and/or additional SMEs are consulted.
Implementation of risk surveys
The analytical risk survey was founded on both a scientific and practical approach for evaluating present and future process control plans and methods. In order to maximize its adoption, the survey workflow was designed to be simple, effective, and easily performed (Figure 3). At a high level it is performed as a two-part process. In the first part, the project team prepares the discussion materials while in the second part, the project team presents to a team of analytical SMEs and discusses the risk survey of the project to generate alignment and possible follow-up activities. The overall workflow includes the following major activities prior to the risk survey committee meeting:
- Holistic analytical controls evaluation: Each project team’s analytical lead (AL) initially evaluates the holistic analytical controls within the context of the API release requirements and CQAs with respect to ICH guideline Q6 and phase of development. All the tests in the analytical controls, including intermediate release and IPCs, are chosen to impart the quality, safety, and efficacy of the drug substance. Relevant data should have been already collected as part of the development and campaign knowledge.
- Triage tests into common risk categories: All testing methods are evaluated and placed into the relevant project risk categories shown in Table 2. The performance history of each method is assessed, and key analytical concerns captured for a project using a templated format. Table 2 provides a list of key analytical categories that are common areas of concern; however, as every project is different, the category population can differ.
- Assessment of tests with performance and gaps: For each individual method, the performance is compared to that expected (see examples in Table 3). Any unusual method conditions, such as uncommon instrumentation, hard to source materials, atypical operating parameters, or parameters near the edge of typical calibrations are specifically evaluated. Where possible, data from the method innovator lab and from other vendor labs is compared to check the method reproducibility. With the above information and the risk band catalog, the risk factor level of each method is standardized to drive consistent evaluation and expectations.
- Collate controls and risks into common document templates: Knowledge gathered from the activities above is used to generate a slide deck from a pre-generated template that includes project background information and risk factors. Banding for each risk characteristic is additionally collated into a spreadsheet template where detailed reasoning of the risk band is explained.
On average, ALs will only need a few hours to collate the requisite information and make or update their template-based documentation (for return visits). Once the AL and their project team have gathered all their required information, the risk survey committee meeting is held. During the meeting, ALs present the assembled risk evaluation using the common document templates to a team of experienced SMEs who have a strong background in analytical method development, validation, and transfer activities. SMEs evaluate the control strategies and the risk banding of each method during the meeting so they may provide their opinions on the project team’s evaluation. SMEs also provide feedback and suggestions to ALs on the control strategies, gaps, method risk banding, and risk mitigation. For all cases where high risks exist, a mitigation plan is required.
The ultimate goal is to ensure alignment between the project team and expert committee on the recommendations and action items, with a commitment for the AL to return with improvements and a revised risk survey in 6 to 12 months if the project is still active. The risk survey meetings are typically 1 to 1.5 hours with approximately 30 minutes presentation time and 30 minutes discussion. The ALs will record all the action items in the spreadsheet comments section. The spreadsheets and presentation decks are stored on a commonly accesible platform, such as a Microsoft SharePoint site.
Risk survey program metrics
So far this program has provided help to more than 40 projects within BMS over 5 years. The collated results of medium to high-risk areas across the portfolio of projects has enabled periodic evaluation by the department leadership. This has helped justify improvements in trainings, SME resourcing, technology and method development workflows across the department and projects.
In Figures 4 and 5, the method performance risk bands identified throughout the year and across many methods are plotted against the risk categories for 2020 and 2021, respectively.
In 2020 the area with the most prevalent high risk was due to analyte degradation either in solution or on the LC column. This degradation may be caused by: intrinsic compound instability; incompatible mobile phases, diluents, or other sample preparation operations; or due to a specific reactive property of the column, such as residual metals. When dealing with such risks, root cause analysis experiments are suggested based upon the data at hand. For example, alternative chromatographic conditions or inert systems can isolate the separation from the sample preparation.
Communication of common liabilities also leads to more awareness of common risks, which allows us to identify opportunities to implement method development strategies to reduce the probability of risk. For example, a workflow change was implemented to reduce the second highest risk contributor in 2020, method selectivity, which was primarily identified in achiral LC methods. An improved LC column screening system was introduced over these two years, as well as a 2DLC main peak purity check to ensure appropriate method selectivity at the earliest stages of development. To address IPC control risks, a training program was implemented to ensure method developers were better equipped to deal with the unique characteristics of IPC methods. These examples highlight how the risk survey program has led to and captured improvements in our performance. However, this analysis also reveals that high and medium level concerns were not completely eliminated and will require more attention moving forward.
Conclusions
Here we have presented a risk survey program applied to the analytical strategy for early to mid- stage development programs. It is a simple assessment on the performance of analytical methods to meet their desired stage-appropriate objectives, and it also serves to capture knowledge and understanding of the method’s merits to improve future development. The risk survey can deliver clear and actionable outputs because it’s driven by evaluating real-world method performance against consistent scientific expectations and criteria. By performing risk surveys across a large and diverse portfolio (>40 projects to date), the program has been able to identify not only concerns within a given project, but also has the additional benefits of driving consistent and improved work practices across the analytical community.
We believe this program can be readily extended beyond small molecule drug substance programs, such as biologics or drug product analytical groups, due to similar quality requirements and analysis techniques, although some modification of categories and banding criteria would be necessary. By using the risk survey process, any analytical team can reduce analytical risk in early development, facilitate stage-appropriate analytical controls, drive inter-project consistency, and identify improvements to method development workflows.
References
- ICH guideline 9 Quality Risk Management
- ICH guideline Q3A Impurities in New Drug Substances
- ICH guideline Q3C Impurities: Guideline for Residual Solvents
- ICH guideline Q3D Guideline for Element Impurity
- ICH guideline M7 Assessment and Control of DNA (Mutagenic) Impurities in Pharmaceutical to Limit Potential Carcinogenic Risk
- ICH guideline Q2 Validation of Analytical Procedures: Text and Methodology
- Peter Tattersall, Qinggang Wang, Li Li and Brent Kleintop, NJCG Symposium, 25 Sep 2019
Author Biographies
Li Li, Ph.D. is currently a Senior Principal Scientist at Bristol-Myers Squibb Company in New Jersey. She received her BSc. at Hunan Normal University, MS from University of Minnesota at Twin Cities, and Ph.D. from Seton Hall University. She previously worked in Analytical Research Department of Merck and Co. at Rahway, NJ. She joined Bristol[1]Myers Squibb in 2005 where she is an analytical team leader within Chemical and Synthetic Development. Her work has been focused on CMC analytical supports for various projects on method development, validation and transfer in support of process development and clinical supply of drug substance synthesis.
Peter Tattersall, Ph.D. is a Director in Chemical Process Development at Bristol-Myers Squibb Company in New Jersey. He received his Ph.D. from the University of Manchester, UK. He previously worked in Analytical Development at AstraZeneca, Wilmington. He joined Bristol-Myers Squibb in 2003 where he is an analytical team leader within Chemical and Synthetic Development. He supervises a small group of analytical chemists working on method development, validation and transfer in support of process development and clinical supply of drug substance synthesis.
Qinggang Wang, Ph.D., is currently a Scientific Director in Chemical Process Development in Bristol-Myers Squibb Company. He received his Ph. D. in Analytical Chemistry from Tsinghua University, Beijing, China, and joined BMS in 2003. His work has been focused on CMC analytical supports for various projects within BMS portfolio, including small molecules, synthetic peptides and oligonucleotides.
Jonathan Shackman is an Associate Scientific Director in the Chemical Process Development department of the New Jersey Product Development group at Bristol Myers Squibb. He received his two BS degrees from the University of Arizona and Ph.D. in Chemistry from the University of Michigan. He previously worked at the U.S. National Institute of Standards and Technology followed as an Assistant Professor of Chemistry at Temple University in Philadelphia, PA. He joined Bristol Myers Squibb in 2010 where he primarily focuses on innovative analytical strategies and technologies to rapidly advance drug development for both small molecule and biologics from pre-clinical through marketed products.
Lianjia Ma, Ph.D. is an Associate Scientific Director in Analytical Division of Chemical Process Development department within Pharmaceutical Development at Bristol-Myers Squibb Company in New Jersey. He received his MS from Iowa State University and Ph.D. from the University of Minnesota at Twin City. He joined Bristol-Myers Squibb in 2005 where he is an analytical team leader within Analytical Research and Development. He worked as Principal Scientist in Celgene from 2018 to 2019 as CMC analytical lead to support the development of drug substance and drug product. He supervises a small group of analytical chemists working on method development, validation and transfer in support of process development and clinical supply of drug substance synthesis.
Adrian Doggett is a Senior Principal Scientist in the Analytical section of Chemical and Process Development at Bristol-Myers Squibb Company in the UK. He received his BSc from Liverpool John Moores University, UK. He joined Bristol-Myers Squibb in 2002. Currently he is a member of a team focusing on method development in support of process development. He has a focus on LC and GC analysis where he is a subject matter expert within BMS.
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!