Abstract
Expiration dating and storage conditions are required on the label for all pharmaceutical products. To establish a drug product’s storage condition and expiration date, a manufacturer must conduct stability studies for all phases of the drug product, from clinical trials to manufacturing and continuing after approval. Stability studies are quality assessment tools to ensure that any chemical, physical, or microbiological changes that may occur as the product ages will not adversely impact the quality of finished products throughout the product’s lifetime. Stability data are also used to establish product specifications to monitor drug product quality, safety, and efficacy. ICH Q1A(R2) is an excellent resource for selecting the appropriate requirements for a stability program; however, ICH is a guideline for new drug substances and drug products; therefore, it is insufficient for acquiring complete knowledge of the stability behavior of a drug product. ICH Q12 has introduced the concept of product lifecycle management, in which post-approval changes are also considered to maintain and improve quality and where knowledge gained throughout the product lifecycle can provide a better understanding of the risks to the stability program. This paper will discuss how the ICH Q12 concept can be built into the stability program and how the Q12 toolbox can connect stability activities supporting product and process changes continuously from the design phase through post-approval, thus fostering continuous improvement for the entire product lifecycle.
Introduction
The ICH Q1A(R2)1 guideline, “Stability Testing of New Drug Substances and Products,” is widely recognized in the pharma industry, outlining stability testing requirements for drug registration. Besides registration, stability testing is also crucial for other stages of drug development. Designing a comprehensive stability program encompassing all phases of drug development can reduce resources, time, and costly redundant testing. ICH Q1A(R2) was initially aimed at the European Community (EC), Japan, and the United States (US); however, many other countries, including Brazil, Mexico, Egypt, Canada, Singapore, Korea, the UK, China, Saudi Arabia, Switzerland, Taipei, and Turkey have since adopted it. The WHO and ASEAN organizations have issued regional guidelines aligning with the ICH Q1A(R2) principles.
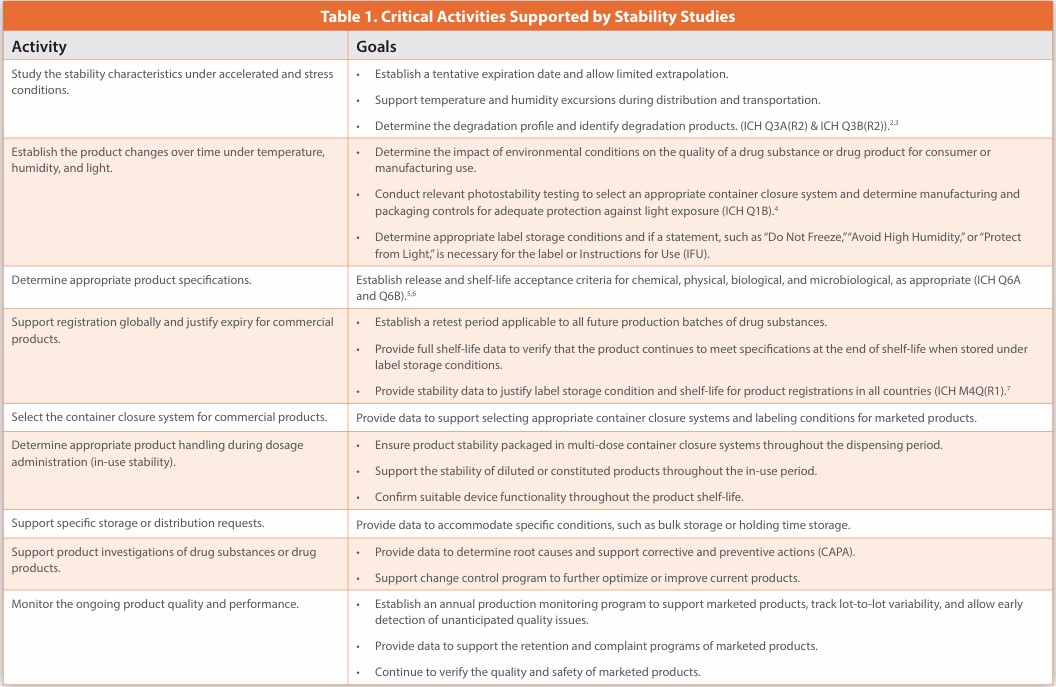
Stability characteristics are critical quality attributes (CQAs) of pharmaceutical products, ensuring a desired quality. As per ICH Q1A(R2), stability testing evaluates how product quality changes over time due to environmental factors, such as temperature, humidity, and light. Failing stability studies post-approval can also lead to product recalls or discontinuation. However, the stability knowledge of a product is not usually linked from development to commercialization as the program is typically developed and transferred in segments. Aside from establishing expiration dates and storage conditions, stability data support various essential activities, as shown in Table 1.
As manufacturers develop complex formulations to meet medical needs, ensuring the quality of active pharmaceutical ingredients (API) and drug products remains challenging. Data integrity and analytical validation are crucial for assessing raw materials and finished products during release and stability. While many publications discuss product and analytical lifecycle management, limited literature addresses designing a robust Stability Program covering the entire product lifecycle and the critical information factors each product phase will bring.8,9,10 This article will attempt to make such a connection. Although the focus is on synthetic small-molecule drugs, many of the concepts can apply to large-molecule biologicals.
Bringing ICH Product Lifecycle Concepts to the Stability Program
On 20 November 2019, ICH finalized Q12,11 providing a framework for post-approval Chemistry, Manufacturing, and Controls (CMC) changes. This concept builds on previous ICH guidelines (Q8(R2), Q9, Q10, Q11)12,13,14,15 to support science- and risk-based approaches in drug development. United States Pharmacopeia (USP) General Chapter <1220> addressed the analytical lifecycle and became official in May 2022.16 Subsequently, ICH revised Q2(R2)17 and published draft Q14,18 recommending implementation on November 1, 2023. These new guidelines facilitate the development and validation of analytical procedures to support product lifecycle and continuous improvement.
Stability evaluation often relies on end-product testing, focusing on regulatory approval rather than product understanding through multi-disciplinary collaboration and prior knowledge. ICH Q12 introduces a comprehensive approach to managing changes across the lifecycle. Applying lifecycle management to stability programs involves setting expiry dates and label storage conditions using data from development to registration and post-approval. Manufacturers can identify risks and develop control strategies to safeguard product quality by building the product and process knowledge into the stability program. This science and risk-based methodology aligns the stability program with the Quality-by-Design (QbD) concept in Q8, as illustrated in Figure 1.

Including a QbD approach in analytical method validation leads to a robust method operable design region (MODR). This region represents the conditions under which the method has been proven to work reliably, ultimately reducing the number of manufacturing investigations due to poor method performance.19 The MODR concept can also be applied to the stability lifecycle, and we can conceptualize a Stability Operable Design Region (SODR) for a global product, where we identify a design space for formulation, process, packaging, and storage conditions to maintain product quality within a targeted expiration date. This concept will be further developed in a forthcoming paper.
ICH Q12 offers a holistic framework for product lifecycle management, comprising four phases: development, transfer, commercial, and discontinuation. Figure 2 shows the activities of the Stability Lifecycle’s four stages, enhancing the understanding of drug substances and product stability characteristics. Many case studies show that lean stability approaches aid the protocol design across clinical, registration, and post-approval phases.20
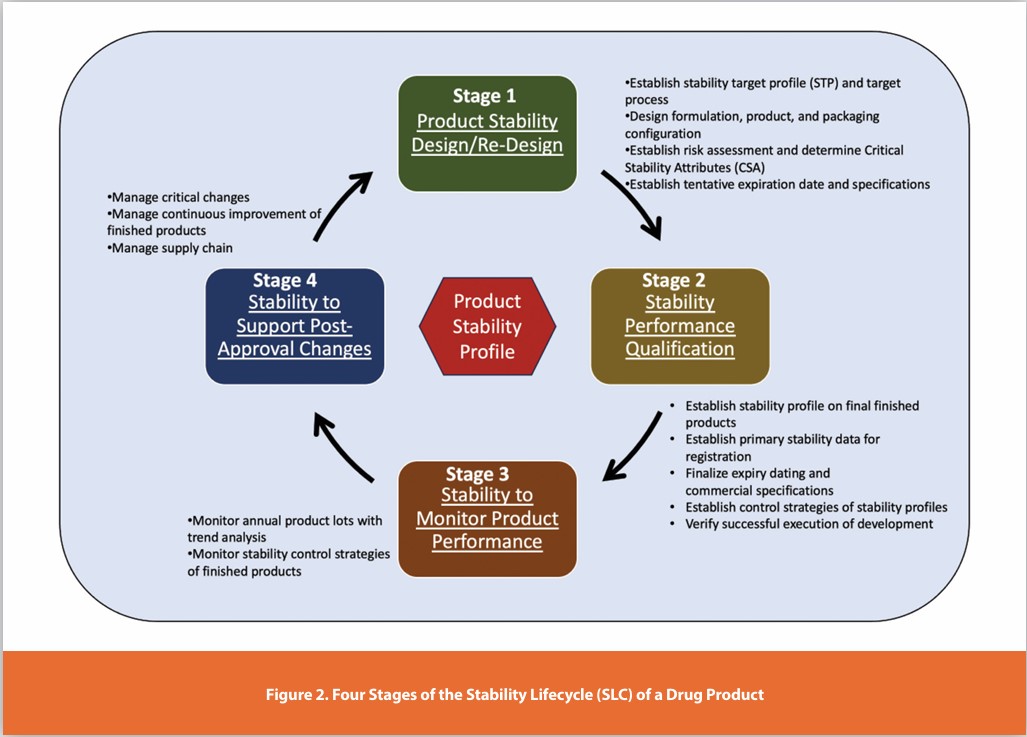
In brief, Stage 1 focuses on establishing the preliminary product stability through clinical materials and establishing the stability target profile and the desired expiration dating period. Stage 2 verifies the critical quality attributes and confirms the stability profile with final formulations for registration. In Stage 3, the annual stability program continuously monitors product quality and safety while adapting to global conditions. Additional studies in Stage 4 support manufacturing changes and distribution while maintaining stability profiles. Integrating stability assessments at each stage enables risk management, resource optimization, and product improvement, avoiding market recalls. The following sections elaborate on the crucial objectives of each stage and provide additional details on the stability lifecycle.
Stage 1 – Stability for Product Design/Redesign
In Stage 1, stability studies support pre-clinical and clinical drug development, generating data for Investigational New Drug (IND) application or equivalent for countries outside the US. Small-scale batches are often available for animal toxicological studies or early clinical phases. Stability protocols are typically short and involve limited time points to establish a stability target profile, critical quality attributes, and preliminary analytical methods. Clinical specifications may also be designed at this stage. Following IND approval, stability studies will continue supporting clinical trials and the development of the commercialization process. A Stability Target Profile (STP), akin to Quality Target Product Profile (QTPP) and Analytical Target Profile (ATP) in Q8 and Q14, guides this stage, ensuring product performance aligns with intended safety and quality throughout its lifecycle. If STP changes, the quality and safety of the drug product should be re-evaluated.
Formulations and packages evolve rapidly in early development, prompting additional studies to support preclinical and clinical configurations. Stability protocols typically match clinical study durations and storage conditions where clinical studies will be conducted. Testing can be extended to gather additional data depending on sample availability. Preliminary accelerated and stress studies are done to understand better how elevated temperature and humidity impact the materials’ stability. This data set also helps develop stability-indicating analytical methods, including non-product-specific testing for physical and microbiological changes.
Critical Stability Attributes (CSA) are determined at this stage, guiding the preliminary establishment of release and stability specifications. These CSA are attributes that can change as the product ages, impacting product quality and safety. Release specifications are often more stringent than stability specifications. However, some regulatory agencies prefer a single set of specifications. Safety studies address impurities exceeding the qualification limit in ICH Q3A, and Q3B.2,3 Risk assessments evaluate the impurity profile and degradation product formation during the clinical phases. Tentative expiration date and label storage conditions are proposed based on available real-time and accelerated stability data, as described in ICH Q1E.21 Additional stability studies will substantiate this tentative expiration date as the program migrates to Stage 2.
Product labels may be developed for different countries; therefore, drug products may have different specifications and expiration dates. Developing submission strategies early on to set up global stability studies is crucial. Lack of planning may cause a delay in submission or redundant stability studies. Predictive stability studies, such as A.S.A.P. or predictive modeling, can provide informed decisions on the drug product’s formulation, packaging, and shelf life.9
This stage includes redesigning a new product or extending a product line due to reformulation, the latest technology, or new API synthesis for marketed products that may face discontinuation. Additional stability studies will be set up based on lessons learned or previous knowledge of product stability.
Stage 2 – Qualification of Stability Performance
Stage 2 of the stability program establishes registration data by conducting additional stability and stress studies on final product formulations using the final manufacturing process and packaging configuration. This stage confirms the critical stability attributes, determines control strategies and sets label storage conditions and shelf-life. Multiple scale-up batches undergo formal stability testing per ICH Q1A(R2) to support Phase III clinical studies and registration. Testing complexity increases due to various marketing packages, strengths, and container-closure configurations. Bracketing and matrixing concepts can be used to reduce analytical testing when appropriate.1 Assuming successful clinical outcomes, the sponsor submits a New Drug Application (NDA) for small molecules or a Biologics License Application (BLA) for large molecules, requesting marketing approval. These primary stability data encompass chemical, physical, biological, and microbiological assessments, guiding label recommendations, shelf-life, and packaging requirements. For global registration of room temperature products in hot and very humid (Zone IVB) regions, like ASEAN or Brazil, a study will be conducted at the 30°C/75% RH condition.

Analytical methods must be established and fully validated in Stage 2, which consists of stability studies supporting Phase III clinical and registration. Specifications at this Stage should also include acceptance criteria for impurities or degradation products. Global stability protocols may assess stability under ICH conditions.22 Stress studies can be repeated under Good Manufacturing Practices (GMP) to confirm the product’s stability profile and specifications. In addition, stress data can also be used to support commercial distribution and transportation. Table 2 provides an example of a section of a formal stability protocol listing assay and impurities testing of a small molecule product.
The labeled storage condition for the above example is room temperature; thus, 25°C/60%RH is the primary condition. The study duration may extend to a year beyond the proposed expiration date but not exceed 60 months (five years). Intermediate (30°C/65%RH) and accelerated (40°C/75%RH) conditions are assessed to establish a tentative shelf-life until real-time data are available. The 30°C/65%RH condition is typically used if significant changes occur at 40°C/75%RH; however, it is unnecessary if 30°C/75%RH conditions are evaluated.
The latter is employed for countries with extreme humidity and temperature conditions, such as ASEAN or ANVISA (Zone IVb).22,23,24
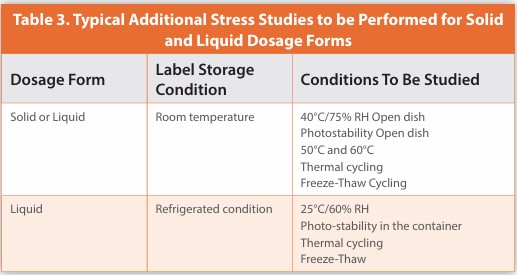
In addition to the above, stress conditions may be applied if there are notable differences in formulation, process, analytical procedures, or packaging compared to previous studies. These conditions should be repeated under GMP in preparation for pre-approval inspection. While one batch suffices for some agencies, others may request stress testing on three submission batches. Table 3 provides typical additional stress conditions for solid or liquid dosage forms. “If degradation products are found in the open-dish condition, samples in the packages should be assessed to ensure the container closure system provides adequate protection in such conditions.
In-use studies are conducted for multidose liquid or solid dosage forms, particularly those sensitive to humidity and temperature variations. These studies may comprise pre- and post-reconstitution, dilution, and compatibility tests for parenteral drugs. Stability programs include upright and inverted configurations to monitor product-container interactions during storage. Special stability studies are conducted for various purposes, such as bulk storage, distribution, and transportation. Water loss should also be considered for liquid products stored in semi-permeable container-closure packages.
Identifying CSA and Critical Process Parameters (CPP) relies on extensive literature searches, prior knowledge, and development stability studies. Statistical and risk assessments per ICH Q913 monitor product stability against the stability target profile (STP)—any critical changes prompt data evaluation for early detection of unintended alterations or product improvement. Risk assessments may be reiterated to confirm the CPP and CSA before NDA or BLA filing.
Control strategies are determined using extensive historical stability data and the body of knowledge (BoK) on the drug product. Out-of-trend (OOT) limits can be established to closely monitor product stability and prevent Out-of-specification (OOS) issues. Stage 2 concludes with finalizing product specifications and expiry based on statistical models, accelerated stability studies, and real-time data from formal stability studies. Development and any historical stability data may be used to support this decision, and a quality risk assessment establishes control strategies for critical attributes. Statistical models and critical attributes influencing product performance on storage guide the development of future stability protocols and post-approval commitments.
Stage 3 – Studies to Monitor Product Performance
Stage 3 follows NDA/BLA submission and approval and involves initiating stability studies using products from the commercial manufacturing site to monitor the marketed product. While Stage 2 studies are conducted on representative batches, the first three Process Performance Qualification (PPQ) batches undergo stability testing with the same protocol to verify the CSA maintenance and monitor product performance, enhancing confidence in batch-to-batch consistency. Control strategies determined in Stage 2 are monitored in Stage 3 for the annual stability program.
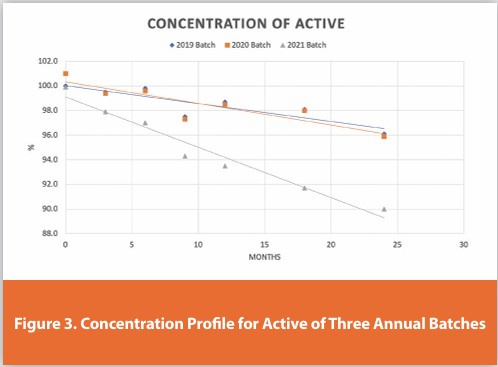
During Stage 3, the product’s expiration date, the final formulation, and packaging specifications are established. Long-term stability testing on required annual production batches confirms batch-to-batch consistency and validates minor material or process changes. Matrix or bracketing schemes are applied depending on production levels to streamline testing. Trend analysis using linear regression identifies unexpected shifts or trends, signaling potential differences in degradation kinetics, primarily oxidation and hydrolysis pathways.25,26 Figure 3 illustrates the stability profile of three annual batches, highlighting a variation that prompts further investigation.
Stage 4 – Studies to Support Post-Approval Changes
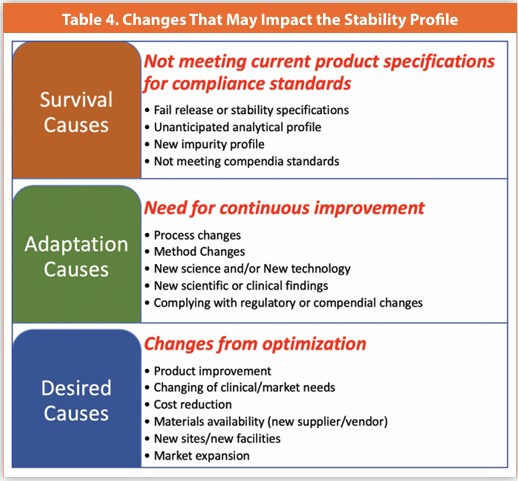
Post-approval changes, driven by survival (stay in business), adaptation (continuous improvement), or desire (consumer or manufacturing optimization), are specific to the drug substance and drug product. Table 4 outlines common reasons for changes that can affect product stability.
A comprehensive risk assessment, drawing on stability knowledge and ongoing monitoring data, is essential to evaluate the impact on stability, safety, and performance. Change control activities require well-defined protocols within the stability program, specifying the number of batches, testing parameters, and duration. Close collaboration among cross-functional teams, such as quality assurance, formulation, packaging engineering, regulatory affairs, and analytical development, is crucial for aligning regulatory requirements and ensuring the submission of regulatory amendments globally.
Science- and risk-based approaches facilitate the effective implementation of changes. Several case studies presented by the IQ Consortium demonstrate leveraging prior product BoK to assess risk and impact, potentially reducing stability data commitments and reporting categories for change implementation as part of post-approval risk mitigation.27,28
Stage 1 introduces STP with the intended shelf-life and storage condition. However, product maturation may necessitate shelf life extension, requiring new stability studies unless this has been accounted for in the initial stability protocol.
Continuous monitoring and trending stability data are crucial for detecting and addressing unexpected trends. Effective communication with regulatory authorities ensures compliance and stability requirements for necessary changes. A robust monitoring system enables systematic data collection, analysis, and interpretation, initiating continuous improvement throughout the product lifecycle.
Increasing raw material costs prompt manufacturers to explore alternative, cost-effective materials without compromising safety or quality. Manufacturing site changes and batch size adjustments are common post-approval changes requiring stability studies to confirm approved shelf-life and storage conditions with new materials. Knowledge and experience gained post-approval drive the scope and design of these stability studies.11 Drug manufacturers must assess changes driven by unexpected supplier alterations, such as discontinuation or substituting raw materials or components, relying on solid product knowledge, including compatibility and comparability testing. Technical expertise in raw material impact and packaging compatibility aids the decision-making process.
Conclusion
The stability lifecycle of a pharmaceutical product, as aligned with ICH Q12, Q2(R2), Q14, and USP General <1220>, requires meticulous management from development through distribution and builds the understanding of the product stability profile. Stability studies provide insights into the product’s behavior under various conditions, steering expiration dates, storage conditions, and transportation requirements. Thus, stability data are crucial to developing control strategies and connecting information from different areas and phases of product development to maintain quality through commercialization. Regulatory bodies determine necessary technical and stability data for global approval, requiring sufficient information to ensure products remain unaffected, emphasizing the synergy between science and compliance. This lifecycle spans four stages that establish the Stability Target Profile (STP), identify Critical Stability Attributes (CSA), and facilitate adjustments in formulation or process as knowledge grows. A holistic risk- and science-based stability program ensures quality and safety throughout a product’s lifespan. It provides a means to strengthen control strategies, effectively manage post-approval changes, and thus mitigate risks to support patient safety.
Acknowledgments:
The authors thank the following colleagues for their time and effort in providing outstanding technical peer reviews: Jing Capucao, Ph.D., GSK; Tage Carlson, Ph.D., Hollister Incorporated; Tony Mazzeo, Ph.D., Bristol Myers Squibb; Lori McCaig, Ph.D., Pfizer; and Hameraj Singh, Ph.D., Pfanstiehl, Inc.
References:
- ICH, Q1A (R2), Stability Testing of New Drug Substances and Products (February 2003).
- ICH, Q3A(R2), Impurities in New Drug Substances (October 2006).
- ICH, Q3B(R2), Impurities in New Drug Products (June 2006).
- ICH, Q1B, Stability Testing: Photostability Testing of New Drug Substances and Products (November 1996).
- ICH, Q6A, Specifications: Test Procedures and Acceptance Criteria for New Drug Substance and New Drug Products: Chemical Substances (October 1999).
- ICH, Q6B, Specification: Test Procedures and Acceptance Criteria for Biotechnological/ Biological Products (March 1999).
- ICH, M4Q(R1), The Common Technical Document for the Registration of Pharmaceuticals for Human Use (September 2002).
- Persich P, Hellings M, Jhajra S, Phalke P, Vanhoutte K. Chapter 5, A Model Approach for Developing Stability-Indicating Analytical Methods. In: Methods for Stability Testing of Pharmaceuticals. Humana Press; 2018.
- Dong MW, Huynh-Ba KC. Stability Studies and Testing of Pharmaceuticals - An Overview. LCGC North America, 38(6), 325-336 (2020).
- Huynh-Ba KC, Holberg W. Analytical Techniques Used in the GMP Laboratory. In: Analytical Testing for the Pharmaceutical GMP Laboratory. Newark, Delaware: John Wiley & Sons, Incorporated; 2022.
- ICH, Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management (November 2019).
- ICH, Q8(R2), Pharmaceutical Development (August 2009).
- ICH, Q9(R1), Quality Risk Management (January 2023).
- ICH, Q10, Pharmaceutical Quality System Q10 (June 2008).
- ICH, Q11, Development, and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) May 2012.
- United States Pharmacopeia (USP) General Chapter, Analytical Procedure Life Cycle. USP-NF. Rockville, MD.
- ICH, Q2(R2), Validation of Analytical Procedures (March 2022).
- ICH, Q14, Analytical Procedure Development Q14 (March 2022).
- Verch, T., Campa, C., Chéry, C.C. et al. Analytical Quality by Design, Life Cycle Management, and Method Control. AAPS J 24, 34 (2022).
- Adamec, E., Babayan, Y., Catacchio, B. et al. Lean Stability Case Studies—Leveraging Science- and Risk-Based Approaches to Enable Meaningful Phase Specific Pharmaceutical Stability Strategies. J Pharm Innov 16, 566–574 (2021).
- ICH, Q1E, Evaluation of Stability Data Q1E (February 2023).
- World Health Organization (WHO), TRS 1010 -Annex 10: WHO guidelines on Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. WHO Technical Report Series 1010, 2018.
- ASEAN Guideline on Stability Study of Drug Product (R1), 25th ACCSQ-PPWG Revision.
- Nacional I. RESOLUTION - RDC No. 318, OF NOVEMBER 6, 2019. RESOLUÇÃO - RDC Nº 318, DE 6 DE NOVEMBRO DE 2019 - RESOLUÇÃO - RDC Nº 318, DE 6 DE NOVEMBRO DE 2019 – DOU (November 2019).
- N. Hartvig and L. Kamper. “A Statistical Decision System for Out-of-Trend Evaluation,” Pharmaceutical Technology 41 (1) 2017.
- Timothy J Snape AA& JD. Understanding the chemical basis of drug stability and degradation. The Pharmaceutical Journal. October 9, 2010.
- McCaig, L., Nowak, S., Abbott, A. et al. Science- and Risk-Based Stability Strategies to Support Product Lifecycle Changes. AAPS J 26, 34 (2024).
- Huynh-Ba K., Editor. Pharmaceutical Stability Testing to Support Global Markets. New York, NY: Springer Science & Business Media; 2010.
Author Details
Kim Huynh-Ba*- Pharmalytik, LLC; Christopher Latoz- Hollister Incorporated. *Corresponding Author
Kim Huynh-Ba is the Managing Director of Pharmalytik Consulting, LLC. Kim is a member of the USP Council of Experts (2015-2025), where she chairs the Small Molecules 4 Expert Committee. Kim is an Adjunct Professor at Temple University-School of Pharmacy and Illinois Institute of Technology (IIT), teaching Quality Audit, ICH quality guidelines, and Good Manufacturing Practices. She is the editor of multiple book volumes, including the “Handbook of Stability Testing in Pharmaceutical Development: Regulations, Methodologies, and Best Practices” (2008), “Pharmaceutical Stability Testing to Support Global Markets” (2010), and “Analytical Testing for the Pharmaceutical GMP laboratory” (2022). She can be contacted at kim.huynhba@pharmalytik.com.
Chris Latoz is Stability Services Manager for Hollister Incorporated, a company that develops, manufactures, and markets healthcare products and services worldwide. Chris has extensive experience in the medical device industry in R&D, materials testing and characterization, and stability. Chris is an American Association of Pharmaceutical Scientists (AAPS) member and serves on the United States Pharmacopeia (USP) Joint Subcommittee for Drug-Device Combination Products. Chris can be contacted at chris.latoz@hollister.com.
Publication Details
This article appeared in American Pharmaceutical Review: Vol. 27, No. 4May/June 2024Pages: 26-33
Subscribe to our newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!