Introduction
Milling is frequently used in pharmaceutical processing to achieve particle size reduction to enhance the bioavailability of poorly soluble active pharmaceutical ingredients (APIs). The intent is to achieve particles with favorable size distributions that enhance dissolution rates due to their increased surface areas. Milling may also be used to obtain a consistent particle size distribution of the API for ensuring content uniformity of the dosage form.
Micronization, the milling operation discussed in this work, is achieved through a high energy air-jet milling process, whereby fast moving API particles are forced to collide, resulting in particle size reduction. Significant attrition is achieved through this process, but significant structural and surface changes are often induced. These changes potentially affect the stability and physicochemical properties of the API, as well as the performance of the formulated product.
Generally, milled materials demonstrate unique physical properties, e.g. crystallization onset temperature, compared to bulk amorphous materials [1-4]. In this work, we characterize amorphous and micronized APIs using differential scanning calorimetry (DSC), solid state nuclear magnetic resonance (SSNMR), gravimetric vapor sorption (GVS), and X-ray powder diffraction (XRPD) to obtain a thorough understanding of the components of the micronization-induced changes.
Characterization of Amorphous APIs
The surface crystallization rates for amorphous solids have been demonstrated to be significantly faster compared to those of bulk amorphous crystallization [5,6]. To further investigate this trend, amorphous griseofulvin was prepared by different techniques, including melt quenching, ball milling, and spray drying, and was analyzed using DSC (see Figure 1a). The melt-quenched sample demonstrates a clear glass transition around 90°C and crystallization onset at 125°C. For the ball-milled material, the only detected transition is crystallization with an onset at 73°C, significantly below the glass transition temperature of the melt-quenched sample. The spray-dried API demonstrates a broad glass transition with a midpoint of 57°C, presumably observed at a lower temperature due to the presence of residual solvent in the sample. Crystallization onset for the spray-dried material is observed at 100°C and the exotherm is bimodal. Bimodal crystallization exotherms have been previously observed for milled griseofulvin.[1,2,4] The peak with a lower onset temperature has been explained in one study as defective crystal rearrangement.[2] In this case, the process of spray drying would not be expected to generate defective crystals, but still clearly shows the same thermal behavior. In yet another study, the explanation for the reduction in recrystallization temperature was suggested to be due to nucleated surface crystallization [1]. Based on the mechanical stress applied during the ball milling process, generation of nuclei at the surface of the material and a bimodal crystallization exotherm were anticipated; however, this thermal behavior was not observed. Additionally, meltquenched griseofulvin was generated with a small number of intimate crystal seeds in the sample. The thermal crystallization of the melt-quenched sample with seeds showed no significant change when compared to a melt-quenched sample without seeds present. What these studies suggest is that the thermal crystallization behavior may be more closely related to the size distribution of the particles, and thus to the degree of surface available for crystallization.
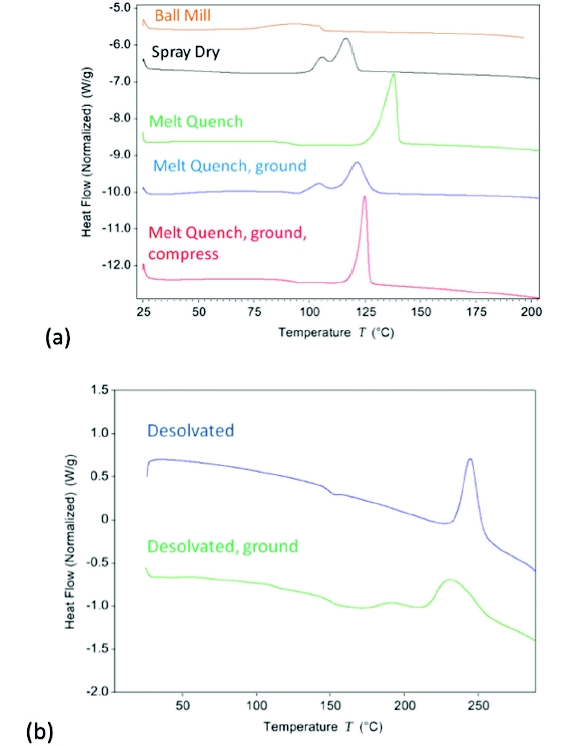 Figure 1. DSC traces for (a) amorphous griseofulvin and (b) amorphous compound A. The eff ect of preparation method on crystallization is noted.
Figure 1. DSC traces for (a) amorphous griseofulvin and (b) amorphous compound A. The eff ect of preparation method on crystallization is noted.To further probe the difference in thermal behavior, the melt-quenched material was gently ground to increase the surface area of the sample. The ground material demonstrated a significant reduction in crystallization onset temperature to 37°C. The sample demonstrated a broad crystallization event over a 100°C temperature range, likely due to the relatively wide particle size distribution obtained with grinding. This transition was trimodal, with a broad shoulder mode below 95°C, followed by a bimodal transition similar to the spraydried amorphous sample. The enthalpies of crystallization obtained for both the ground and non-ground samples are comparable, at ~85 J/g. The ground material was subsequently compressed in a Carver press to revert to a lower surface area. The compressed sample demonstrates a clear glass transition around 90°C and crystallization onset of 117°C, recovering thermal properties similar to the initial low surface area melt-quenched sample (refer to Figure 1a). These results are consistent with the literature [1] and clearly demonstrate that the energy required to achieve crystallization is significantly lower for high surface area amorphous griseofulvin. The effect of surface area on the thermal behavior of amorphous materials was further evaluated using compound A. Amorphous compound A was generated by desolvation at 200°C, forming a molten sample that was subsequently cooled to ambient temperature. The high surface area amorphous sample was obtained by trituration of the desolvated material. The glass transition for the desolvated compound A is 147°C and onset of crystallization is around 234°C, as shown in Figure 1b. The crystallization event for the desolvated ground sample is observed over the range of about 100-260°C, with onset below the glass transition temperature of the non-ground amorphous sample. Similar thermal behavior was observed for both systems, where the higher surface area amorphous samples demonstrated crystallization onset below the glass transition of the lower surface area samples, presumably due to enhanced mobility at the surface [6].
SSNMR was used to confi rm the increase in molecular mobility for high surface area amorphous materials. 13C NMR was used to characterize amorphous griseofulvin and 19F NMR was used to characterize amorphous compound A. High and low surface area amorphous samples were analyzed for both compounds. The nuclear spin-lattice relaxation time, T1, and spin-lattice relaxation in the rotating frame, T1ρ, are generally expected to be shorter for molecules with higher mobility since they would return to equilibrium more rapidly. The T1 and T1ρ values are consistent with higher mobility in the high surface area samples demonstrated by shorter relaxation times, as shown in Table 1.
Table 1. T1 and T1ρ values obtained from 13C and 19F NMR analysis for amorphous griseofulvin and compound A, respectively.
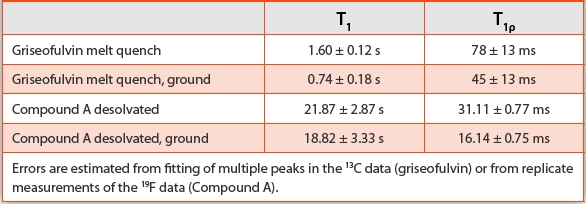
The thermal and NMR results are consistent with an increase in mobility at the surface of the amorphous materials. The eff ect of surface area on molecular mobility and crystallization is confi rmed by the reversible thermal properties of the amorphous materials, as well as the relaxation times obtained from the NMR analyses.
Characterization of Micronized Apis
The high energy particle attrition achieved during micronization is expected to generate disorder that generally resides on the surface of the particle and demonstrates high surface area. The characterization of micronization-induced disorder can often be challenging due to the small amount that is formed relative to the predominantly crystalline particle. In the following section, DSC, GVS, SSNMR and XRPD are used to examine the surface and structural changes associated with process-induced disorder.
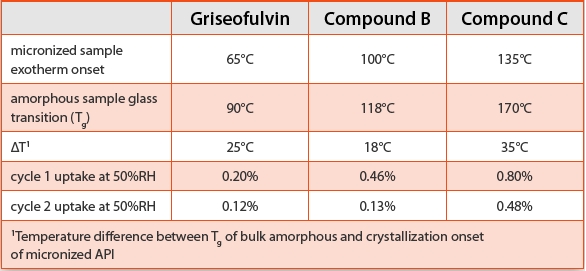
The DSC traces for three diff erent crystalline micronized APIs all exhibit small exothermic events with onsets below the bulk glass transition temperature known for each compound, as shown in Figure 2. Since compound A is a solvate, a similar assessment of the crystallization of micronized API could not be completed due to the desolvation event in the DSC trace. The exothermic transitions observed for the micronized samples are consistent with a small amount of amorphous content generated on the surface of high surface area micronized particles. As described above, the onset of crystallization for high surface amorphous content would be expected to be well below the glass transition of bulk amorphous. It is noted that changes to processing parameters, such as micronization energy, may shift the observed onset or affect the magnitude of the exotherm for the micronized samples. Regardless of the processing conditions applied, the onset for all of the micronized systems studied was observed below the glass transitions observed for bulk amorphous materials generated with a range of surface areas from various techniques. The micronized APIs were also analyzed using GVS, where the total sorption includes surface adsorption as well as absorption attributable to the presence of disordered material. Exposure to moisture may initiate crystallization, which results in the expulsion of bulk absorbed water molecules and reduction in the overall quantity sorbed at a particular relative humidity condition. The samples were exposed to two isothermal cycles up to 90%RH and for all the systems, the second sorption cycle demonstrated less moisture uptake compared to the initial cycle. Thermal analysis of the micronized samples following the GVS analysis showed significant reduction in the magnitude (compound C) or the absence (griseofulvin and compound B) of the crystallization event, consistent with rapid crystallization at the surface of the particles. Compound C demonstrates the highest crystallization onset temperature, and not surprisingly, the disordered phase is more stable compared to the other micronized materials. When compound C was processed with lower micronization energy, the crystallization exotherm was not observed following a single GVS cycle. The DSC and GVS results are consistent with the proposal that the micronized samples have high surface area and high mobility amorphous regions that require less energy to crystallize compared to bulk amorphous materials. The presence of amorphous phase in micronized compounds A and C was confirmed using SSNMR, however, it was not detected in micronized griseofulvin or compound B. The technique may not be able to detect the low levels of amorphous phase in these samples or the conditions applied during analysis may have initiated crystallization, consistent with the DSC and GVS results that showed griseofulvin and compound B require less energy to recrystallize. In fact, crystallization of ground amorphous griseofulvin was observed during SSNMR studies.
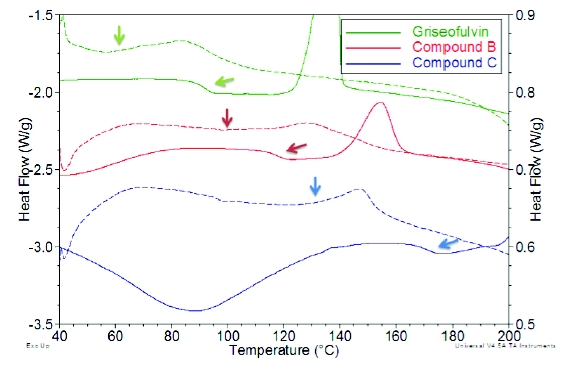 Figure 2. Comparison of DSC traces for bulk amorphous (solid lines) and micronized (dashed lines) APIs. The arrows indicate the Tg of the bulk amorphous and crystallization onset for the micronized samples.
Figure 2. Comparison of DSC traces for bulk amorphous (solid lines) and micronized (dashed lines) APIs. The arrows indicate the Tg of the bulk amorphous and crystallization onset for the micronized samples.Table 2. Summary of DSC and GVS Results for Micronized APIs
Elevated baselines in XRPD patterns are often an indicator of the presence of amorphous content, however, the micronized systems showed no evidence of an elevated baseline. The XRPD peak widths are related to the average crystallite size in a powder sample, and can be used to study structural changes [7]. It is noted that crystallite size and particle size are different; a particle is made up of single crystals separated by fractures/grain boundaries defined as crystallites. Once the average crystallite size falls below a certain size range (typicallyIn order to obtain high quality data to use in the line width calculations, capillary samples were collected via XRPD transmission and the peak line width (FWHM) calculations were made using Rietveld refinementn techniques. The calculated line widths for the micronized APIs are signifi cantly broader relative to the non-micronized input materials, as summarized in Table 3. In systems where the material was processed using diff erent micronization energies, a relationship between the peak broadening and the output particle size was noted.
Table 3. Calculated XRPD Line Widths (FWHM) for Non-micronized and Micronized APIs
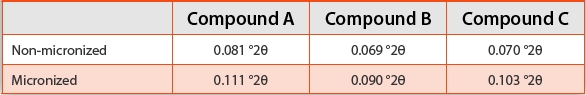
Since signifi cant diff erences were observed between the nonmicronized and micronized samples, line width analyses provide suffi cient space to probe changes to the crystallite structure as a function of temperature and humidity. Compound C was exposed to diff erent stressed environmental conditions and analyzed to examine the eff ect of annealing on the micronized sample. Figure 3 shows the resulting XRPD line widths as a function of time at three humidity conditions when stored at 20°C and 50°C. At both temperatures, annealing was observed, demonstrated by sharpening of the XRPD line widths. After one week, signifi cant annealing was observed, and by the second week, the trend appeared to have leveled off . The high relative humidity samples (~95%RH) for both temperatures showed the most signifi cant annealing. The micronized sample stored at 50°C/95%RH for two weeks showed a line width approaching 0.08 °2θ, which is not as narrow as the non-micronized line width, 0.07 °2θ, but demonstrates significant annealing and possible increase in crystallite size in a relatively short time period. The broadening of the calculated XRPD line width was consistent with the generation of nanocrystalline content during micronization and the phase annealed with an apparent synergy of temperature and relative humidity. Further work is required to determine if strain is also contributing to the peak broadening and to the changes observed in the annealed samples. Additional work is ongoing to correlate these findings to changes observed between freshly micronized and aged APIs that have been stored under typical warehouse conditions.
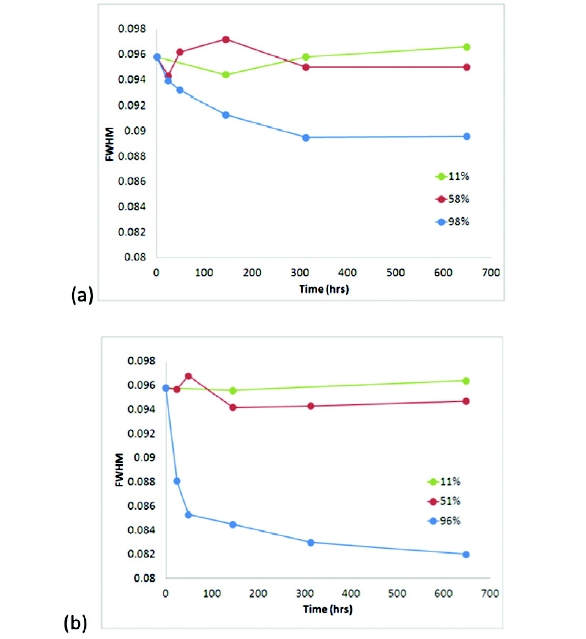 Figure 3. Calculated XRPD line widths (FWHM) for micronized compound C exposed to diff erent environmental conditions over time: (a) 20°C (b) 50°C.
Figure 3. Calculated XRPD line widths (FWHM) for micronized compound C exposed to diff erent environmental conditions over time: (a) 20°C (b) 50°C.The thermal and vapor sorption properties of the micronized samples exposed to different environmental conditions were also tested at various time points. The crystallization events attributable to high mobility surface amorphous phase and the line width sharpening due to annealing of the nanocrystalline phase occurred on different time scales. The crystallization of the amorphous content occurred at a significantly faster rate compared to the annealing of the nanocrystalline content, indicating that different components of disorder are generated during micronization. Correlations between changes in the surface area and porosity of the micronized samples and the XRPD line width analysis are being investigated to further characterize the nanocrystalline content in these systems.
Conclusions
The crystallization onset temperature of amorphous APIs is influenced by surface area and SSNMR was used to demonstrate a correlation between molecular mobility and surface available for crystallization. Based on the physical characteristics of micronized APIs, it appears that there are different components of disorder that are generated as a result of the mechanical stress incurred during processing. DSC, GVS, and SSNMR results are consistent with the formation of surface amorphous regions with enhanced molecular mobility. XRPD line width analyses suggest the formation of structural changes consistent with nanocrystalline content. The annealing of the nanocrystalline phase of micronized systems can be monitored using XRPD line broadening analysis. Work is ongoing to develop a comprehensive understanding of the individual components of disorder on the physicochemical properties and stability of micronized APIs.
References
- N.S. Trasi, S.X.M. Boerrigter, S.R. Byrn, “Investigation of the milling-induced thermal behavior of crystalline and amorphous griseofulvin” Pharm Res 27 (2011)
- T. Feng, R. Pinal, M.T. Carvajal “Process induced disorder in crystalline materials: differentiating defective crystals from the amorphous form of griseofulvin” J Pharm Sci 97 (2008)
- K.J. Crowley, G. Zografi “Cryogenic grinding of indomethacin polymorphs and solvates: assessment of amorphous phase formation and amorphous physical stability” J Pharm Sci 91 (2002)
- S. Chattoraj, C. Bhugra, C. Telang, L. Zhong, Z. Wang, C. Sun “Origin of two modes of nonisothermal crystallization of glasses produced by milling” Pharm Res 29 (2012)
- L. Zhu, J. Jona, K. Nagapudi, T. Wu ”Fast crystallization of griseofulvin below Tg” Pharm Res 27 (2010)
- T. Wu, Y. Sun, N. Li, M.M. deVilliers, L. Yu “Inhibiting surface crystallization of amorphous indomethacin by nanocoating” Langmuir 23 (2007)
- P. Scherrer, “Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen,” Nachr. Ges. Wiss. Göttingen 26 (1918)
- E.A. Van Arkel “Deformation of the crystal lattice of metals by mechanical working” Physica 5 (1925)
- G.K. Williamson, W.H. Hall “X-ray line broadening from filed aluminium and wolfram” Acta Metall (1953)
Author Biographies
Rachel G. Forcino, Ph.D., is an investigator within Product Development at GlaxoSmithKline. Her interests include solid state analysis and process development of pharmaceutical materials. Specific focus has included the characterization of crystalline and amorphous materials using thermal analysis, x-ray diffraction, and physisorption techniques.
Jeff Brum, Ph.D. received his Ph.D. in physical chemistry from Tulane University. He started with GlaxoSmithKline in 1994, and is currently team leader of the physical properties group in Collegeville PA. Prior to joining GSK he was a National Research Council Postdoctoral Fellow at the National Institute of Standards and Technology.
Glenn R. Williams, Ph.D. is interested in solid-state characterization of amorphous and crystalline pharmaceutical materials (active drug substances, excipients, and drug products). In particular, he has been focused on x-ray diffraction, x-ray fluorescence, microscopy, thermal analysis, and molecular spectroscopy. He has over 12 years of solid-state analysis experience in the pharmaceutical industry from both the brand and generic perspectives.
Frederick G. Vogt, Ph.D. is a patent attorney at Morgan, Lewis & Bockius, LLP. His scientific interests include NMR, x-ray diffraction, and vibrational and molecular spectroscopy, as applied to drug substance crystallization and form control, drug product and excipient characterization and secondary manufacturing processes, and patent support. He has more than 10 years of experience in these fields and has contributed to more than 50 peer-reviewed publications and five book chapters involving spectroscopy and solid-state analysis.