Introduction
In June 2013, the European Medicines Agency (EMA) approved a realtime polymerase chain reaction (Real-Time PCR) Mycoplasma test for lot release of matrix-applied characterized autologous cultured chondrocytes (MACI®). MACI® is an autologous cell therapy product indicated for the repair of articular cartilage defects in the knee in the European Union (EU). Implementing rapid microbiological methods (RMM) for lot release of cell therapy products with short shelf lives improves the safety of these products and ultimately leads to increased protection for patients. Mycoplasma species are the smallest known free-living organisms, ranging in size from 0.15 to 0.3 μm in diameter. They are self-replicating bacteria that lack cell walls and are relatively common contaminants of mammalian cell culture [1] in a research setting. Mycoplasma present particular challenges as they are difficult to culture and detect using traditional microbiological detection techniques. Conventional test methods for Mycoplasma detection [2-4] take at least 28 days to complete. Developing a rapid method to detect Mycoplasma was necessary for lot release of MACI®, which has a 6-day shelf life. Selecting a method based on Real-Time PCR facilitated successful development, validation, and technology transfer. The test takes less than six hours to complete; automation of sample preparation steps further reduces that time to five hours. Implementing the newly approved method in a production environment provided an excellent opportunity to reflect on the validation and technology transfer process.
Methodology Overview
The sample configuration for the Real-Time PCR Mycoplasma test is the same as for the conventional growth-based test. It contains drug product in conditioned cell culture medium. Centrifugation pellets any Mycoplasma organisms present in the sample and concentrates them to improve detection. After lysing the organisms and enzymatically digesting proteins that could interfere with nucleic acid recovery, an extraction with proprietary magnetic particlesand a magnet collects and purifies any DNA in the sample. Final preparation of the DNA extract requires elution of the DNA and removal of the magnetic particles. Using a Real-Time PCR Instrument, the assay analyzes the DNA extract using optimized multiplexed primer design for specific and comprehensive Mycoplasma species detection. Positive samples exhibit a threshold cycle (Ct) inversely proportional to the amount of DNA present and an amplicon melting temperature (Tm) in a specific range. The assay also includes a Discriminatory Positive/Extraction Control, which is a large plasmid containing a Mycoplasma DNA sequence. The Tm of the Discriminatory Positive/Extraction Control is different than the Tm of Mycoplasma DNA, eliminating the possibility of a false positive test result due to accidental cross-contamination of a test sample with control DNA. The assay uses a negative control, positive control, extraction control, and inhibition controls to ensure the validity of test results.
Table 1. Validation Study
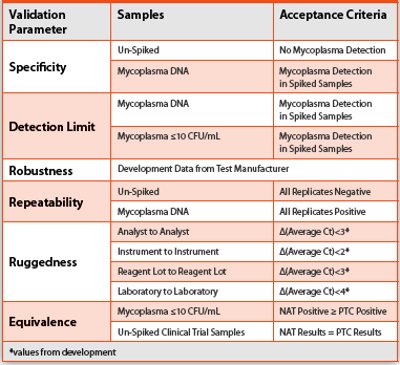
The Real-Time PCR Mycoplasma testing procedure complies with PhEur 2.6.7 Mycoplasmas, which specifies requirements for validation of a nucleic acid-based test [4]. The validation study tested drug product samples derived from multiple chondrocyte strains spiked with viable organisms (and corresponding DNA solutions) from six Mycoplasma species. This method is a qualitative test for the presence or absence of microorganisms, similar to a sterility test for bacteria and fungi. Specificity and Detection Limit are the most critical validation parameters for this type of qualitative test. USP <1223> Validation of Alternative Microbiological Methods and PhEur 5.1.6 Alternative Methods for Control of Microbiological Quality specify additional validation parameters for a qualitative test including Robustness, Repeatability, and Ruggedness [5, 6] (ICH has not yet addressed alternative microbiological methods). PhEur 2.6.7 also requires an assessment of Equivalence, since the Real-Time PCR method is a replacement for the conventional culture method [4]. Table 1 contains a summary of the validation study design.
Lessons Learned – Validation
Overall, the validation experiments went smoothly given the complexity of the study and the logistics of execution. Even so, it is important to look for opportunities for continual improvement when working in a cGMP production environment. This validation presented three such opportunities in the areas of reference standards, logistics, and detection criteria.
Reference Standards
Preparing reference standards containing a well-characterized quantity of correctly-identified viable Mycoplasma organisms requires specialized techniques [7]. Commercial sources of Mycoplasma organism reference standards characterized in terms of their genomic DNA copy to colony forming unit (GC/CFU) ratio [8] are currently available from multiple suppliers. Determining the GC/CFU ratio is necessary to establish whether an alternative nucleic acid test yields results equivalent or better than a compendial method. It is important to reduce the nucleic acid contribution from non-viable organisms because DNA detection is not necessarily equivalent to viable organism detection. Culture methods detect only viable organisms, while the Real-Time PCR test detects Mycoplasma DNA from both viable and non-viable sources. With knowledge of the GC/CFU ratio, it is possible to establish the detection limit solely using purified Mycoplasma DNA reference standards, which are also now commercially available. Neither Mycoplasma organism reference standards characterized in terms of GC/ CFU nor purified DNA was available for the validation study conducted in 2011.
Table 2. Mycoplasma Solution GC/CFU Ratio
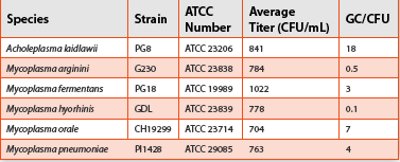
Overcoming this hurdle required working closely with a contract laboratory specializing in growth and detection of Mycoplasma. The laboratory used stock solutions from organisms harvested during log-phase growth to maximize viability. They prepared viable Mycoplasma organism solutions at a titer of approximately 1000 CFU/mL from these stock solutions and confirmed the actual titer by plate count. They also prepared non-viable lysed organism solutions (unpurified DNA) from the viable organism solutions equivalent to 1000 CFU/mL. The validation report calculated the GC/CFU ratio from calibration curves correlating number of genome copies (GC) and threshold cycle (Ct) based on an average Mycoplasma genome size of 1,000 kbp. Table 2 contains the average titer and GC/CFU ratio for each species used in the validation study.
The low GC/CFU ratios provided additional supporting data indicating that the enumerated organism stock solutions obtained from the contract testing laboratory and used for spiking were of high viability (not biased toward PCR) and were appropriate for use as calibrated reference standards to compare the performance of the Real-Time PCR test with the performance of the official method. Although this approach yielded favorable results in this case, use of wellcharacterized reference standards is preferable when designing and executing validation studies.
Logistics
Introducing viable Mycoplasma cultures into an autologous cell therapy manufacturing facility producing thousands of lots per year presented an unacceptable contamination risk. Most cell culture facilities choose to eliminate any risk from test lab personnel contaminating production cultures by banning viable Mycoplasma cultures in the entire facility, including the test lab. This presented significant logistical challenges for validation. Performing the portion of the validation protocol requiring use of viable organism reference standards off -site was the most straightforward solution. Working closely with the test manufacturer identified a one-week window in which to perform this portion of the study in their non-cGMP training facility, three-thousand miles away. Considerable challenges related to equipment, assay materials, Mycoplasma reference standard solutions, and training existed.
First, the equipment in the facility was not part of a cGMP metrology program and subject to routine calibration. The equipment used in this validation fell into the following categories: Real-Time PCR Instrument, centrifuges, heat blocks, pipettors, and timers. The Real- Time PCR Instrument was the most critical piece of equipment used in the validation. Executing an Installation Qualification Protocol and Operation Qualification and Instrument Performance Verification Protocol during the week prior to performance of the validation experiments mitigated the risk associated with using the Real-Time PCR Instrument because the protocols confirmed that the instrument was operating within its tolerance range during the validation period. For the remaining equipment, assessing the risks of false positive and false negative results from the use of out-of-tolerance equipment found that assay controls in place mitigated these risks.
Second, interaction of computerized purchasing systems initiated shipments for all of the assay materials from the test manufacturer to the development laboratory, only to ship back to the test manufacturer training facility upon receipt. The reference standards were viable Mycoplasma organism solutions requiring storage at -80°C. Obtaining these solutions at the training facility without compromising the cold chain required coordinating the interactions of the contract manufacturer and test manufacturer as a third party to ensure appropriate storage at all stages (initial storage at contract manufacturer, shipping onC, timely receiving, and storage at the training facility).
Finally, training for the analyst performing the validation experiments occurred in the development laboratory using familiar equipment. Executing the validation study in a non-cGMP training facility using unfamiliar equipment turned out to be a significant challenge. Adding a time constraint to experiments comfortably completed in a longer period only added to the stress. As it happened, the analyst successfully completed the experiments in the time available by executing multiple experiments simultaneously.
Again, while the approach yielded favorable results, the ideal situation would have been to execute the entire validation in the development laboratory. When this is not possible, using an off -site cGMP lab with adequate time for training, material qualification, and protocol execution is preferable.
Detection Criteria
Original detection criteria for positive samples subjected all samples exhibiting an amplicon having a Tm in the correct range for Mycoplasma to additional testing in a conservative attempt to minimize the number of false negative results. Analysis of spiked media samples and samples from reference chondrocyte cultures confirmed this as being a feasible approach to take into validation. During validation for equivalence, however, analysis of 78 retain samples from a clinical trial of an autologous cell therapy revealed baseline noise in the melting curve having a Tm in the correct range for Mycoplasma. Testing using the conventional growth-based method had previously found these samples to be negative for Mycoplasma.
The baseline noise occurred sporadically in one of the duplicate test wells, resulting in false positive results in one well and negative results in the other well. This conflicting data interpretation resulted in an invalid test rate of 29%, which was too high to be useful for lot release of autologous cell therapy products, requiring final results on the same day as product assembly.
A statistical analysis predicting an equal percentage of false positive results and false negative results provided a rational Ct value to include in the results interpretation criteria for determining positive results. The analysis independently confirmed the Ct cutoff recommended by the test manufacturer in updated product documentation. Using the new criteria reduced the invalid test rate from 29% to 5%, which was reasonable for routine testing. The revised interpretation criteria required an assessment of the potential impact to the validation results. After reanalyzing all of the validation results, the report determined that using the revised interpretation criteria did not impact the results of the validation.
Although this situation was not ideal, it provided invaluable insight into the capabilities of the assay prior to implementation. Though not always feasible, these results recommend introducing as much sample variability into the assay development process as possible, especially for individualized treatments such as autologous cell therapy products.
Lessons Learned – Technology Transfer
Technology Transfer of the assay from the development laboratory in the United States (sending unit) to the production facility in Europe (receiving unit) occurred during the regulatory approval process after successful validation.
Technology Transfer and ongoing training faced similar challenges to validation logistics due to banning viable Mycoplasma cultures in the entire production facility, including the test lab. As a result, Technology Transfer and training needed to find a substitute to demonstrate the ability of the analysts to recover organisms without using viable Mycoplasma. Demonstrating proficiency in the full assay procedure was not possible using Discriminatory Positive/Extraction Control (DNA plasmid) spikes alone because the first step in the sample preparation involves centrifugation of an aqueous cell culture medium sample. Since DNA is soluble in water, it will primarily reside in the supernatant after centrifugation and will go undetected by the assay. Fortunately, the test manufacturer uses Escherichia coli (E. coli) to express the Discriminatory Positive/Extraction Control plasmid and they will provide this E. coli strain to their customers. The assay will not detect E. coli DNA, but will detect the Mycoplasma DNA sequence in the plasmid. Using this E. coli, which centrifuges similarly to Mycoplasma, it was possible to develop a working E. coli suspension for spiking at approximately 10 CFU/mL to mimic the assay detection limit and verify each analyst’s performance at that level.
After solving the technical issues surrounding demonstration of proficiency, there were also some positive lessons from the Technology Transfer exercise. First, it was critical to eliminate conflicting priorities at both the sending and receiving units prior to initiating the study. Second, performing intensive analyst training at the sending unit in the US, away from the daily routine at the receiving unit in the EU,reduced the amount of time required for training and allowed focused time to develop analyst expertise.
Conclusion
Rapid detection of Mycoplasma is essential for autologous cell therapy products with short shelf lives. Although product contamination is infrequent, the ability to detect potential contaminants prior to implantation is a critical component of product safety to ensure patient protection. This is likely one reason that final product testing for Mycoplasma is a regulatory requirement for cell therapy products. Developing, validating, and transferring a rapid Mycoplasma test based on Real-Time PCR presented many unique challenges. Validation challenges were either logistical, related to banning viable Mycoplasma cultures in the production facility, or technical, related to appropriate reference standards and detection criteria. Technology Transfer challenges related to demonstrating proficiency given the constraints on use of viable cultures. Proactively addressing each challenge was critical to successful validation, technology transfer, and implementation as a routine lot release test.
Author Biography
 John Duguid is a Principal Process/Analytical Scientist at Genzyme, a sanoficompany, responsible for developing and implementing rapid microbiological assays. Previously, he managed QC cell therapy operations. Mr. Duguid received his BS in Chemistry from the University of Michigan. Prior to Genzyme, he worked as an analytical chemist at Abbott Laboratories and Arthur D. Little.
John Duguid is a Principal Process/Analytical Scientist at Genzyme, a sanoficompany, responsible for developing and implementing rapid microbiological assays. Previously, he managed QC cell therapy operations. Mr. Duguid received his BS in Chemistry from the University of Michigan. Prior to Genzyme, he worked as an analytical chemist at Abbott Laboratories and Arthur D. Little.
References
- FAQ. Bionique® Testing Laboratories, Inc. Web site. 2013. Available at: http://www.bionique. com/. Accessed August 13, 2013.
- Recommended Procedures for Detection of Mycoplasma Contamination in Biological Products Produced in Cell Substrates. Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals. Rockville, MD: The United States Food and Drug Administration Center for Biologics Evaluation and Research; 2003.
- <63> Mycoplasma Tests. In: United States Pharmacopeia. 33rd ed. Rockville, MD: The United States Pharmacopeial Convention, Inc.; 2010.
- 2.6.7. Mycoplasmas. In: European Pharmacopoeia. 6th ed. Strasbourg, FR: European Directorate for the Quality of Medicines; 2010.
- <1223> Validation of Alternative Microbiological Methods. In: United States Pharmacopeia. 32nd ed. Rockville, MD: The United States Pharmacopeial Convention, Inc.; 2009.
- 5.1.6. Alternative Methods for Control of Microbiological Quality. In: European Pharmacopoeia. 6th ed. Strasbourg, FR: European Directorate for the Quality of Medicines; 2010.
- Duguid J. Top Ten Validation Considerations when Implementing a Rapid Mycoplasma Test. Am. Pharm. Rev. 2010; 13:26-31.
- Dabrazhynetskaya A, et al. Collaborative study report: Evaluation of the ATCC experimental mycoplasma reference strains panel prepared for comparison of NAT-based and conventional mycoplasma detection methods. Biologicals 2013. Available at: http://dx.doi.org/10.1016/j. biologicals.2013.07.002. Accessed August 13, 2013.