Introduction
Non-sterile drug Products Contain Microorganisms at the End of Manufacturing.
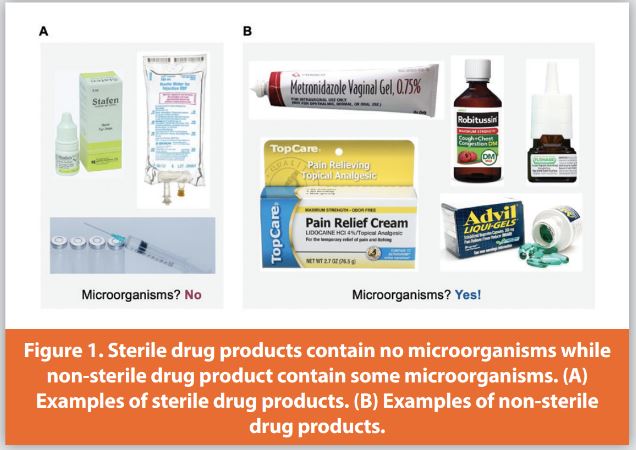
To a pharmaceutical microbiologist, there are two categories of finished pharmaceutical products: non-sterile and sterile. At the end of manufacturing, no microorganisms are present in sterile products. Examples of sterile finished dosage forms are injections, intravenous fluids, and ophthalmic drops (Figure 1A). In contrast, at the end of manufacturing non-sterile drug products, some microorganisms may be present. Examples of non-sterile finished dosage forms are solid oral tablets, nasal sprays, oral liquids, and topical creams (Figure 1B).
Manufacturers mitigate microbial contamination in finished drug products by intentionally designing the drug formulation, the manufacturing processes, and the finished product testing to consider the microbial quality of the finished drug product. To control microbial contamination in products, manufacturers can formulate the drug solution with antimicrobial properties, restrict the bioburden of incoming materials, incorporate bioburden-reducing and sterilization steps during manufacturing, and test the finished product at release and during stability storage (Figure 2).
Non-sterile drug products are tested to determine the number and the type of microorganisms present using USP <60>, USP <61>, and USP <62> test methods to meet USP <1111> acceptance criteria.

At the end of manufacturing non-sterile drug products, manufacturers test the finished product to determine the number and the type of microorganisms present in the finished drug product. To determine the appropriate acceptance criteria for the number and the type of microorganisms present in the finished non-sterile drug product, manufacturers and regulators refer to USP <1111> Microbiological Examination of Nonsterile Products: Acceptance Criteria for Pharmaceutical Preparations and Substances for Pharmaceutical Use. Table 1 in USP <1111> lists acceptance criteria for microbial limits and specified microorganisms for non-sterile dosage forms based on the intended route of administration of the non-sterile drug product. Depending on the manufacturing processes and other risk factors, such as the patient population and route of administration, further limits on total microbial quantity and specified microorganisms may be necessary in addition to the acceptance criteria stated in USP <1111>. For example, aqueous dosage forms are also expected to be tested for the absence of Burkholderia cepacia complex (BCC).

To test finished non-sterile drug products for microbial limits and specified microorganisms, manufacturers use the USP test methods USP <60> Microbiological Examination of Nonsterile Products – Tests for Burkholderia Cepacia Complex, USP <61> Microbiological Examination of Nonsterile Products: Microbial Enumeration Tests, and USP <62> Microbiological Examination of Nonsterile Products: Tests for Specified Microorganisms. To incorporate these USP test methods into routine testing of drug products, the methods must first be demonstrated to be suitable for testing the specific drug product (also referred to as “method suitability”) (Figure 3).
The USP test methods are demonstrated to be suitable for the drug product being tested by introducing indicator microorganisms in the presence of the drug product, performing the test procedures, and assessing the recovery rate of the indicator microorganisms.
Method Suitability Begins With “Preparation of the Sample” Described in USP <61>
The first step of assessing the suitability of the method is the “preparation of the sample” (see first bullet under “Suitability of the Method in the Presence of Product” in Figure 3). USP provides detailed instructions on how to prepare samples of product for microbial limits and specified microorganisms for the following types of drug products:
- Water-soluble products
- Nonfatty products insoluble in water
- Fatty products
- Fluids or solids in aerosol form
- Transdermal patches
But what if the product does not fit into any of these categories?
USP <61> states at the beginning of the “preparation of the sample” section of the chapter:
The method for sample preparation depends on the physical characteristics of the product to be tested. If none of the procedures described below can be demonstrated to be satisfactory, a suitable alternative procedure must be developed.
In this article, a method to develop a “suitable alternative procedure” to prepare samples of novel drug products is proposed.
Developing a “Suitable Alternative Procedure” to Prepare Samples for Microbial Limits and Specified Microorganisms Testing
To prepare samples of novel drug products for microbial limits and specified microorganisms, USP states a “suitable alternative procedure” must be developed based on the physical characteristics of the product to be tested. However, USP does not define how to develop an alternative procedure that meets the criteria of “suitable,” leading to the following questions:
- How are the physical characteristics of the drug product defined?
- How is an alternative procedure demonstrated to be “suitable”?
- Are there published standards and/or guidance documents to reference which can provide guidance?
ISO 11737-1:2018 Describes Methods for Sampling Products for Bioburden Based on Physical Characteristics of the Product
Looking to published standards, ISO 11737-1:2018 Sterilization of health care products – Microbiological methods – Part 1: Determination of a population of microorganisms on products describes how to select a method for sampling products for bioburden based on the physical characteristics of the product. ISO 11737-1:2018 offers the following guidance:
1. A decision tree containing questions on the physical characteristics of the product with links to methods of sampling products based on the product’s physical characteristics is provided in Figure A.1 of ISO 11737- 1:2018.
2. Once a method to sample products is selected based on the physical characteristics of the product, the total processing time of the sampling step must be defined. ISO 11737-1:2018, B.2.1.7 recommends limiting the processing time to minimize changes in microbial population.
3. The selected sampling method and processing time can be demonstrated to be appropriate and suitable for the product being sampled by determining the bioburden recovery efficiency. ISO 11737-1:2018, Annex C describes the “inoculated product” approach to determining bioburden recovery efficiency. This method involves inoculating the product with ~ 100 colony-forming units (cfu) of bacillus spores, performing the sampling method at the proposed processing time, and calculating the percent recovery efficiency of Bacillus spores by dividing the spores recovered in the test sample by the spores recovered in the positive control sample. Percent recovery efficiencies at or near 100% demonstrate the sampling method and processing duration are appropriate and suitable to recover bioburden from the product.
Once an alternative procedure for sampling the novel drug product is defined and demonstrated to be suitable, the alternative sampling procedure can be used to prepare samples for USP <60>, USP <61>, and USP <62> method suitability and, ultimately, routine testing of non-sterile drug products.
Case Study: Developing a Suitable Alternative Procedure to Sample a Novel Drug/Device Combination Product for Microbial Limits and Specified Microorganisms Testing
The approach described above from guidance in ISO 11737-1:2018 was used to develop a suitable alternative procedure for sampling a novel drug/device combination product for microbial limits and specified microorganisms testing. The novel drug/device combination product consists of a nasal swab device with drug solution absorbed in the foam tip of the nasal swab device (Figure 4A and Figure 4B). The nasal swab combination product does not fit into any of the five categories described in the “preparation of the sample” section of USP (i.e., it is not a water-soluble product, nonfatty product insoluble in water, fatty product, fluid or solid in aerosol form, or transdermal patch) and, therefore, a suitable alternative procedure must be developed based on the physical characteristics of the novel nasal swab drug/device combination product.
Selecting the Sampling Method of the Novel Nasal Swab Combination Product
According to ISO 11737-1:2018, Figure A.1 According to the decision tree in ISO 11737-1:2018, Figure A.1, “stomaching or shaking followed by filtration/plating” are appropriate sampling methods based on the physical characteristics of the novel nasal swab drug/device combination product (Figure 4C).
Demonstrating the Sampling Method of the Novel Nasal Swab Combination Product is Suitable
Shaking on an orbital shaker for 0, 5, 15, or 30 minutes f, followed by filtration, was selected as the sampling method to test and compare in bioburden recovery efficiency studies (Figure 5A). Nasal swabs were inoculated with ~ 100 cfu B. subtilis spores, inoculated swabs were placed in media, and then the swabs in media were shaken for 0, 5, 15, or 30 minutes. At the end of the shaking processing time, the media from the swabs in media samples was filtered in duplicate, filters were plated on agar plates, agar plates were incubated, and any colonies recovered on agar plates were counted. Positive controls were performed by inoculating media directly with ~ 100 cfu B. subtilis spores and processing samples as described for test samples. The average recovered inoculum in test and positive control samples was calculated by averaging the two duplicate plates for each sample. Percent spore recovery efficiency was calculated by dividing the test average recovered inoculum by the positive control average recovered inoculum and multiplying by 100. Percent bioburden recovery efficiency was demonstrated to be similar and at or near 100% for all four tested sampling methods (Figure 5B).
A 5-minute shake followed by filtration was selected to prepare samples for USP, USP, and USP method suitability and, ultimately, routine testing of the non-sterile, novel nasal swab drug/device combination product.
Conclusion
To prepare samples of novel products for microbial limits and specified microorganism testing, develop a “suitable alternative procedure” based on the physical characteristics of the product to be tested using the following steps:
- Use ISO 11737-1:2018, Figure A.1 Decision Tree to define the sampling method of the product based on the physical characteristics of the product.
- Limit the processing time of product sampling to minimize changes in the microbial population.
- Demonstrate the sampling method is “suitable” by determining the bioburden recovery efficiency.
References
- United States Pharmacopeial Convention. Chapter Microbiological Examination of Nonsterile Products—Tests for Burkholderia cepacia Complex. United States Pharmacopeia and National Formulary (USP-NF). 42nd ed. Rockville, MD: United States Pharmacopeial Convention; 2019.
- United States Pharmacopeial Convention. Chapter: Microbiological Examination of Nonsterile Products—Microbial Enumeration Tests. United States Pharmacopeia and National Formulary (USP-NF). 42nd ed. Rockville, MD: United States Pharmacopeial Convention; 2019.
- United States Pharmacopeial Convention. Chapter Microbiological Examination of Nonsterile Products—Tests for Specified Microorganisms. United States Pharmacopeia and National Formulary (USP-NF). 42nd ed. Rockville, MD: United States Pharmacopeial Convention; 2019.
- International Organization for Standardization. ISO 11737-1:2018: Sterilization of Health Care Products—Microbiological Methods—Part 1: Determination of a Population of Microorganisms on Products. Geneva, Switzerland: International Organization for Standardization; 2018.
Author Details
Julia Marré, PhD, Regulatory Affairs Principal Consultant, NSF
Julia Marré is a regulatory affairs professional at NSF with prior senior-level experience as a manufacturing and microbiology reviewer at the FDA and as the regulatory lead for numerous drug, biologic, and combination products. She is an expert in assessing the microbiological quality, process, and facilities of a pharmaceutical manufacturing process. She currently serves as an Expert Advisor on the US Pharmacopeia Drug-Device Combination Products Joint Subcommittee and the planning committee for the Parenteral Drug Association’s Pharmaceutical Microbiology Conference.
Publication Details
This article appeared in American Pharmaceutical Review:
Vol. 28, No. 1
Jan/Feb 2025
Pages: 26-29
Subscribe to our e-newsletters.
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!