James Agalloco- Agalloco & Associates
Introduction
EMA’s Annex 1, Manufacture of Sterile Medicinal Products, includes expectations for the manufacture of sterile products. 1 However, the Annex essentially equates the performance of different aseptic technologies, which has the effect of slowing the implementation of advanced aseptic technologies that support improvements in product/patient safety. Caution and confusion abound concerning the use of isolation technology. There is minimal appreciation for their benefits concerning existing practices. A trio of sentences provides EMA’s minimal support for advanced technologies:
“The use of appropriate technologies (e.g. Restricted Access Barriers Systems (RABS), isolators, robotic systems, rapid/alternative meth ods and continuous monitoring systems) should be considered to increase the protection of the product from potential extraneous sources of endotoxin/pyrogen, particulate and microbial contami nation such as personnel, materials and the surrounding environ ment, and assist in the rapid detection of potential contaminants in the environment and the product” (sec 2.1).
“Restricted Access Barrier Systems (RABS) or isolators are beneficial in assuring required conditions and minimizing microbial contamination associated with direct human interventions in the critical zone. Their use should be considered in the CCS. Any alternative approaches to the use of RABS or isolators should be justified.” (sec 4.3)
“Where possible, the use of equipment such as RABS, isolators, or other systems should be considered to reduce the need for critical interventions into grade A and to minimize the risk of contamination.” (sec 8.9)
It must be noted that EMA considers barrier systems, including RABS (with limited further distinction) and isolators, as equally capable. This perspective denigrates the proven superior performance and the acknowledged preference for isolators over even the most sophisticated of RABS designs. 2-4 Moreover, Annex 1 mandates practices for isolators that are more rigorous than those imposed on RABS designs. (sec 4.19-4.21). As the document makes little or no distinction between the various types of RABS, some of which are only marginally more capable than the ordinary manned cleanroom, the incentive for real advancement of aseptic processing technology is sharply reduced. There are added cautions with respect to isolator usage that exceed those for less capable designs, e.g., isolator gloves are subject to intensive testing requirements, while there is no mention of integrity testing for gloves installed in RABS. (sec 4.21)
Given our disappointment with the available draft, several experienced sterile product experts, including the author, called for EMA to address the weaknesses in Annex 1’s treatment of aseptic technology in February 2022. An excerpt from that letter follows:
“Manned cleanrooms were first introduced in the 1950s, and unenhanced, these are functionally obsolete. Considering the best interests of patients across the globe, we believe it is appropriate for EMA to propose and establish a termination date for the use of manned aseptic cleanrooms of five years from the date of publication of the Annex. RABS, a derivative technology that provides performance improvements, would be granted an additional five years of use. RABS technology can be used as a relatively low-cost upgrade of manned cleanrooms, allowing for a more gradual replacement of manned environments.” (see sidebar for original letter)
EMA was not open to this suggestion, considering it problematic from a product supply perspective and its applicability to a broad array of processes/products. Nevertheless, EMA includes other problematic elements in Annex 1.
“Particular attention should be given when the adopted product sterilisation method is not described in the current edition of the Pharmacopoeia, or when it is used for a product which is not a simple aqueous solution. Where possible, heat sterilisation is the method of choice.” (sec 8.37)
This short paragraph challenges the in-situ sterilization of direct and indirect product contact parts with any of the typical agents used for isolator preparation. This hurts many existing and future facilities using separative technologies, making their use more complex. This subject will be expanded upon later in this article.
The following paragraph delayed the implementation of a mandate for lyophilizer sterilization by a year from the effective date of Annex 1 (25 August 2023) to allow firms to either rectify or justify their practices concerning lyophilizer sterilization. That this might have detrimental consequences to product supply was ignored!
“Lyophilizers and associated product transfer and loading/unloading areas should be designed to minimize operator intervention as far as possible. The frequency of lyophilizer sterilisation should be determined based on the design and risks related to system contamination during use. Lyophilizers that are manually loaded or unloaded with no barrier technology separation should be sterilised before each load. For lyophilizers loaded and unloaded by automated systems or protected by closed barrier systems, the frequency of sterilisation should be justified and documented as part of the CCS.” (sec 8.123)
Overall, Annex 1 deems all prevalent technologies for aseptic processes (cleanrooms, RABS, and isolators) as acceptable for continued use and thus of suitable performance. The superiority of isolator technology in assuring sterility and/or containment over less certain manned environments, including Restricted Access Barrier systems (RABs), is widely accepted across our industry. The required presence and allowed access of aseptically gowned personnel in the less capable RABS and manned cleanrooms are considered essentially equivalent to the far superior isolator. The elimination of contamination from the aseptic operator has been pursued for decades to remove the primary source from the critical environment. There is little justification for a standard that essentially equates a known contaminated environment with one that approaches true sterility in operation. Mandating a zero contamination rate in environments with different capabilities does not support their operational equivalence! (sec 9.30)
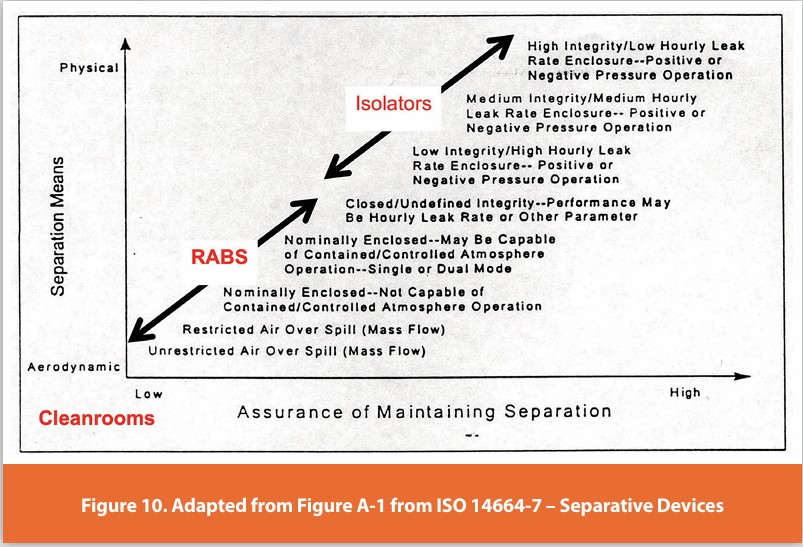
A review of the meaningful differences across aseptic processing technologies is provided in Table 1 and described/visually depicted in this narrative (see Figures 1-9). These images use a dirty cleanroom technician to indicate shedding contamination. These images serve as a visual reminder of the reality that aseptically gowned and qualified operators continuously shed both microorganisms and particles 5,6 The first images (Figures 1-3) depict older designs, which should be considered functionally obsolete. Many of these are still in use, and surprisingly, there are some in various stages of installation. In the latter images (Figures 7-9), the operator need not be aseptically garbed, as the near absolute separation of personnel from the critical environment prevents the introduction of contamination. It must be recognized that no system that allows operator access is truly protected from contamination ingress. The descriptions and images should be understood as generic examples, and the details of any individual system vary and may include elements associated with different systems. The overall hierarchy of systems shown here is consistent with ISO 14644-7, Cleanrooms and Associated Controlled Environments — Part 7: Separative devices, which describes cleanroom technologies concerning the type of separation, the means for confirming the separation, and the effectiveness of the separation.7 Figure 10 adds an overlay to Figure A.1 from the ISO14644-7 standard to highlight the significant performance distinction between manned cleanrooms, RABS, and isolators. As RABS do not maintain a defined pressure differential between critical surfaces and personnel, they cannot control human-derived contamination as effectively as isolators. Only in their most evolved state do CRABS design approach isolator-like capability, but even there, they fall short. It is important to note that manned cleanrooms fall outside the Figure 10 image entirely unless one considers a hinged door or flexible curtain a meaningful form of separation. Manned cleanrooms lack real separation between gowned personnel and critical surfaces and perform less capably than even the least capable RABS design.
Conventional Manned Cleanroom
Figure 1
The very first cleanrooms provided little or no physical separation between personnel and the critical zone, relying almost entirely on unidirectional air, aseptically garbed personnel, and good aseptic technique to protect sterile materials. All activities must be performed by the operator. The operator is in an ISO 5/6 environment supplied with non-unidirectional air.

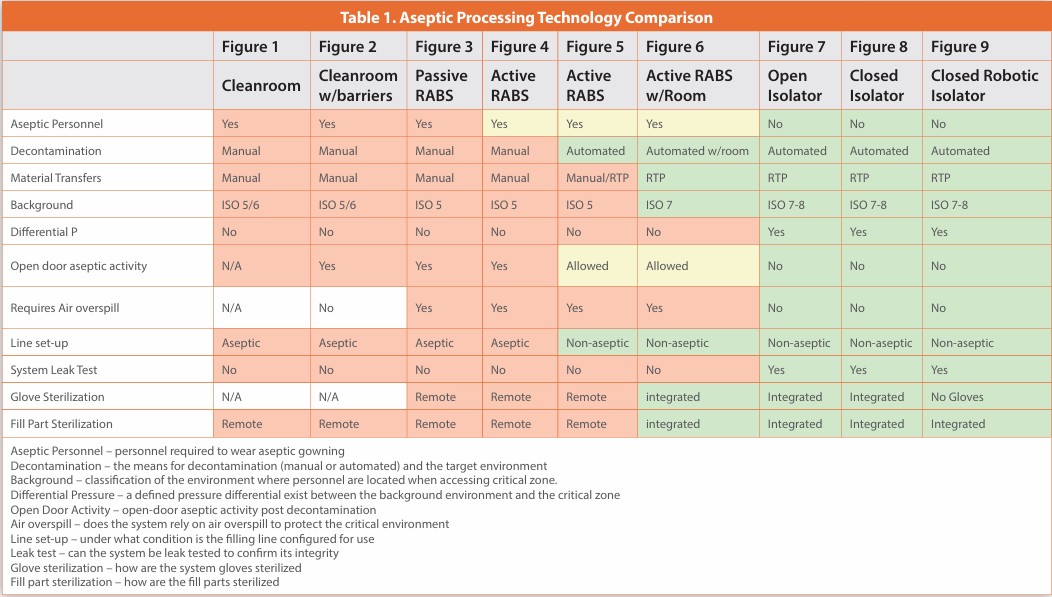
Manned Cleanroom with Barriers
Figure 2
A common refinement to the manned cleanroom was the addition of rigid barriers separating the operator(s) from the critical zone. These barriers may be safety interlocked with the internal machinery to pause its operation while open. These barriers are opened to allow the operator unimpeded access for set-up, and the conduct of inherent and corrective interventions. The operator is in an ISO 5/6 environment supplied with non-unidirectional air.
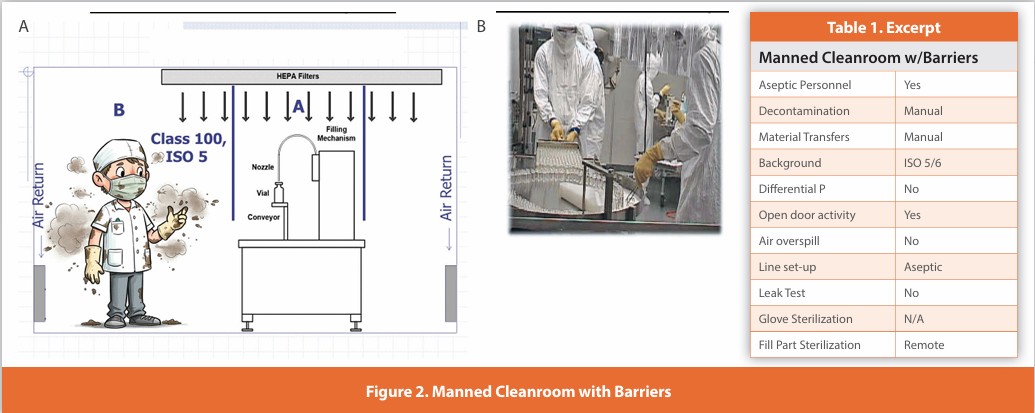
Passive RABS
Figure 3
A separative design where sterilized gloves are affixed to the enclosure surrounding the critical zone. There is no separate air supply to the enclosure other than the HEPA-filtered supply to the room. These systems are typically manually decontaminated; line setup and glove installation are performed aseptically. Depending upon the specific intervention, doors may be opened. Component addition may or may not be performed without opening the enclosure. Added unidirectional air outside the RABS is rare.
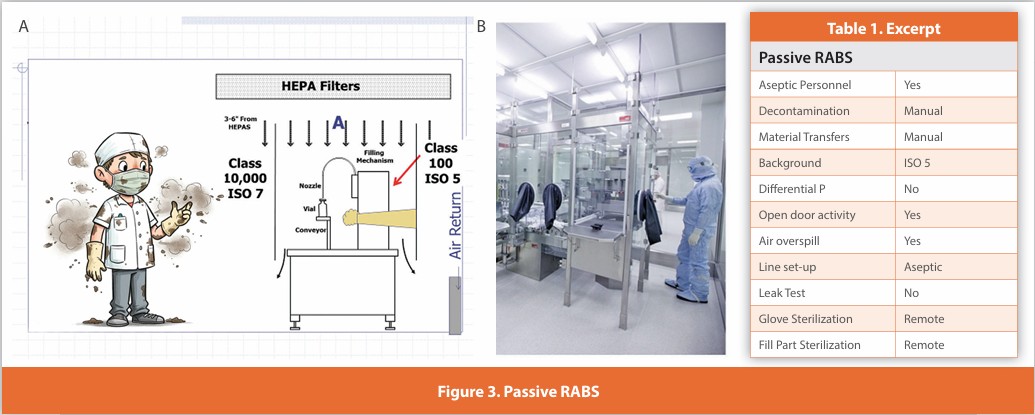
Active RABS (manually decontaminated)
Figure 4
A separative design where sterilized gloves are affixed to the enclosure surrounding the critical zone. Air within the enclosure is HEPA filtered using air either drawn from the room or directly from the air supply. These systems are manually decontaminated; line set-up and glove installation are performed aseptically. Depending upon the specific interventional activity, doors may be opened perhaps less frequently than with passive RABS. Component addition is more likely to be performed without opening the enclosure. When the doors are open, the operator is located within an external unidirectional air supply (not shown in the photograph).
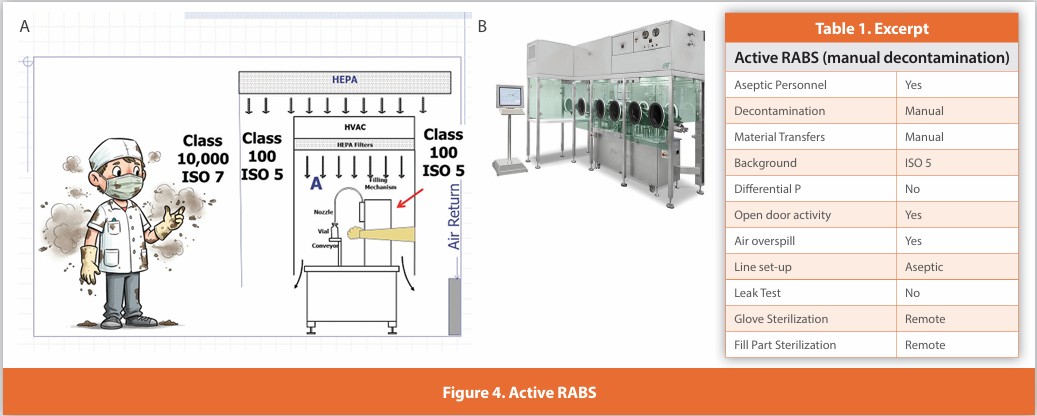
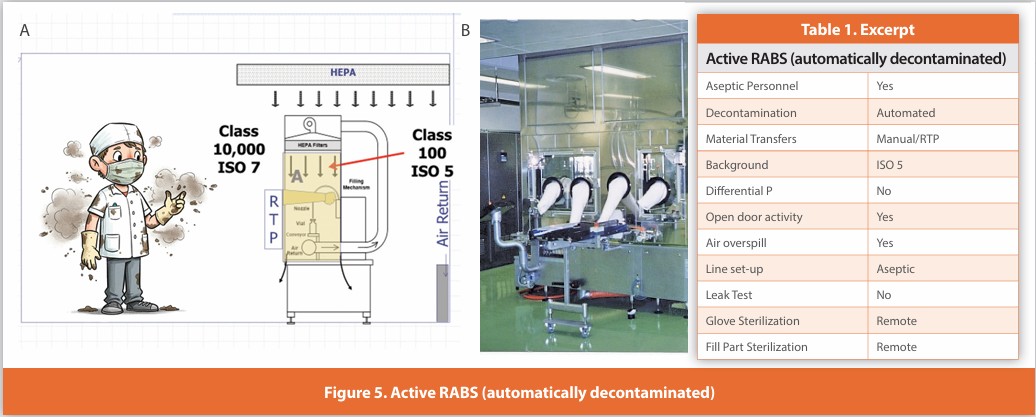
Active RABS (automatically decontaminated)
Figure 5
A separative design where gloves are affixed to the enclosure surrounding the critical zone. Air within the enclosure is HEPA filtered using air either drawn from the room or directly from the air supply. Line set-up and glove installation are performed non-aseptically before decontamination. These systems are automatically decontaminated while closed; the system then relies upon air overspill to protect the critical zone during operation. Component addition is performed without opening the enclosure using isolator-like methods, as the enclosure is not opened during operation. The described design is not in wide usage.
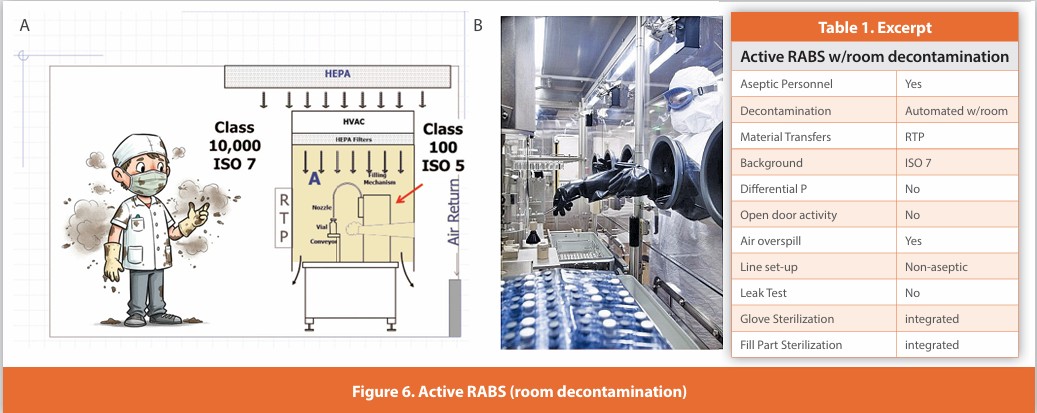
Active RABS (room decontaminated)
Figure 6
A separative design where gloves are affixed to the enclosure surrounding the critical zone. Air within the enclosure is HEPA filtered using air either drawn from the room or directly from the air supply. Line set-up and glove installation are performed non-aseptically before decontamination. The room and RABS are simultaneously automatically decontaminated; the system then relies upon air overspill to protect the critical zone during operation. Component addition is performed without opening the enclosure using isolator-like methods, as the enclosure is not opened during operation. The described design is not in wide usage.
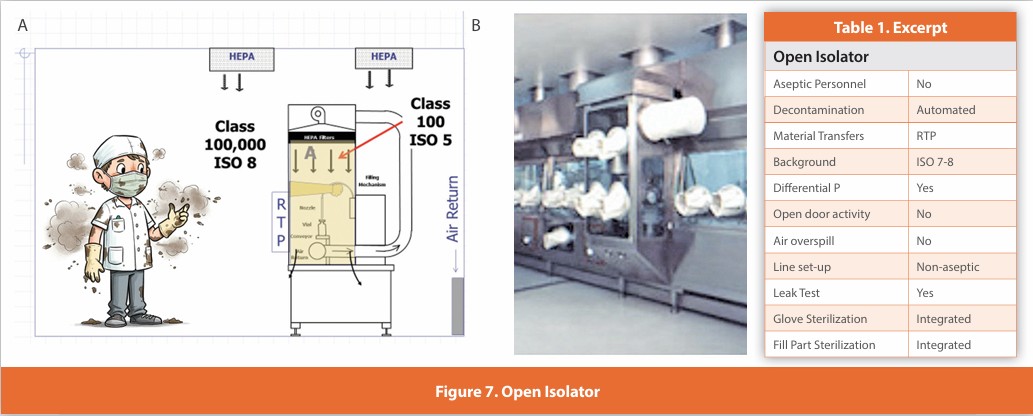
Open Isolator
Figure 7
An isolated design where gloves are affixed to the enclosure surrounding the critical zone. Air within the isolator is HEPA filtered using air drawn from the room. Line set-up and glove installation are performed non-aseptically, and the isolator is automatically decontaminated. Component addition is performed without opening the isolator using engineered systems for continuous ingress/egress. The isolator maintains positive pressure during operation and is never opened during use. This design is in widespread use. Air overspill at the filled container exit.
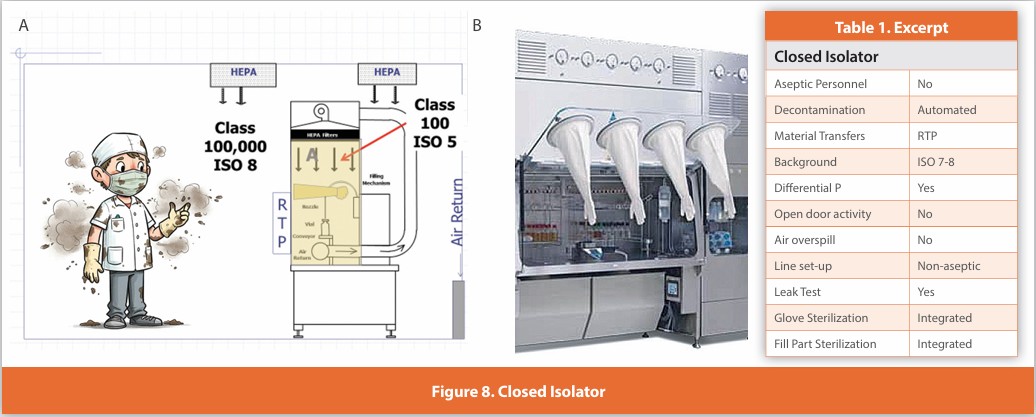
Closed Isolator
Figure 8
An isolated design where gloves are affixed to the enclosure surrounding the critical zone. Air within the isolator is HEPA filtered using air drawn from the room. Line set-up and glove installation are performed non-aseptically, and the isolator is automatically decontaminated. Component transfer is performed without opening the isolator using specifically designed batch transfer systems for ingress/egress. The isolator maintains positive pressure at all times and is never opened during use. This design is in use globally for batch operations such as cell processing, API manufacturing, clinical or small-scale manufacturing, as well as for research.
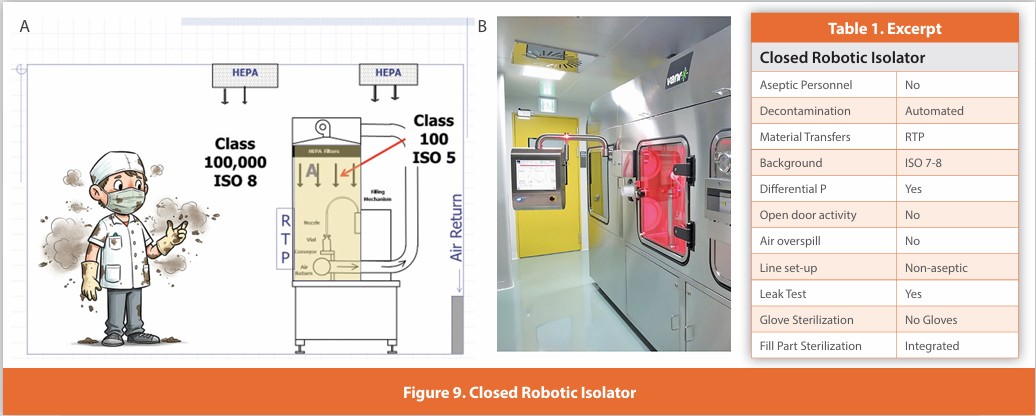
Closed Robotic Isolator
Figure 9
An isolated design where no gloves are required. All internal activities are managed by either robotic or machine automation. Air within the isolator is HEPA filtered. Line set-up is performed non-aseptically, and the isolator is automatically decontaminated. Component movement is performed without opening the isolator using integrated batch transfer systems for ingress/egress. The isolator maintains positive pressure at all times and is never opened during use. This design is in expanding usage, especially for smaller batch sizes.
A Continuum Isn’t Constant
Many of these systems can appear similar in appearance; however, there are substantial differences in how they are operated that are critical to their performance and make a singular narrative inappropriate. Respect that individual designs vary, and features can vary from those described above. Consider the following important features:
- Is the assembly of the line done aseptically by gowned personnel? Systems requiring aseptic assembly do not adequately separate personnel from critical equipment and surfaces. Any design requiring that invasive interventional activity is no better manned cleanroom.
- Is the opening of the enclosure necessary during use? Opening of the enclosure at any time during processing compromises the barrier’s integrity. Systems where that is necessary provide inadequate separation of gowned personnel from sterile materials.
- Is the system manually decontaminated? Reliance on gowned personnel to decontaminate the critical environment is less reliable than automated treatment. Locations can be missed, and re-contamination can occur.
- Can the system be leak tested? The adequacy of a separative device is affirmed by the ability to conduct a periodic leak test to confirm the integrity of the designed safeguards. Systems that cannot be leak tested increase contamination risk.
- Is the system protected by air overspill? Reliance on air flow to protect sterile material surfaces is less certain than the use of a controlled and alarmed pressure differential.
- Are interventions with enclosure gloves required? The elimination of gloves removes the most vulnerable component of many separative designs.
The answers to these questions reveal the true capabilities of the system (see Table 2).
The presence of a “barrier” between the operator and critical surfaces does not establish its adequacy for aseptic processing. Considering very different systems under the broad term “barrier technologies” when they differ substantially suggests an interchangeability/equality that does not exist. The most capable represent “state-of-the-art” designs, while the least capable may be only marginally better than a manned cleanroom. What makes separative technologies better than a manned cleanroom is the elimination of direct inherent and corrective interventions by aseptically gowned personnel.8 Many RABS systems fail to do that. Some of the cautionary statements in the Annex 1 draft relate to the least capable designs, which, when applied to the most capable systems, compliance-driven complexity without any reduction in patient risk (sec 4.5-4.17)
RABS are described as systems where personnel access to the critical environment is “restricted” by their configuration and operating practices; however, they are only truly advanced and equal in performance to isolators if they eliminate all human intervention during ssetupand processing 8 The difference between “restricted access” and “no access” is infinite; therefore, any RABS design that allows operator access for direct intervention in ISO 5 is substantially more rrisk-intensivethan any isolator. It’s ludicrous to even mention the lesser RABS configurations in the same context.9,10
Annex 1 substantially overstates the performance of RABS, relying heavily on characteristics of the most capable systems to describe their operation. While RABS with these features are in operation, they represent only a fraction of the installed base of RABS worldwide. 3 There are substantially more RABS designs in use that not only allow direct operator intervention but also cannot operate without it. Annex 1 fails to recognize that RABS encompasses a variety of different configurations and that, with few exceptions, the majority of RABS offer only minimal improvements over manned cleanrooms.9 The authors of Annex 1 appear unable to discern the significant difference in risk mitigation between truly advanced systems and those that remain aseptic operator intervention dependent. This is an egregious error because it encourages firms to maintain and even continue to install less capable designs with the belief that they are equal in performance according to the Annex (see Figure 10). The best systems automate everything from setup to lyophilizer loading and eliminate manual interventions. This is not a trivial difference; it supports major advances in performance and patient safety.
The Decontamination/ Sterilization Conundrum
Influenced by biological indicator failures experienced in some isolator systems, Annex 1 considers vapor phase hydrogen peroxide processes decontamination as something less than sterilization. In this, it echoes an earlier position from MHEW on the limitations of this process.11 The net effect of this is to require that product contact parts be subjected to a validated sterilization process other than vapor phase H2O2 transferred, and then installed aseptically within a decontaminated isolator system. This requirement has impacted investment in isolator-based systems and placed numerous pre-existing lines in limbo. There are weaknesses in EMA’s position considering vapor phase H2O2 as being other than a sterilizing agent.
- The outdated restraint imposed by EMA’s position on sterilization processes, “Particular attention should be given when the adopted product sterilisation method is not described in the current edition of the Pharmacopoeia …“ (sec 8.37). As EP’s sterilization content was last revised in 2005, it lags well behind other standards, making technological advances more difficult.12
- Numerous validated and fully licensed manufacturing facilities use vapor H2O2 routinely for sterilization of product and non-product contact parts. Conversion to the expected EMA approach is not feasible in many of these installations due to equipment size, equipment weight, and access/configuration limitations 3,13
- In 2024, the US Food and Drug Administration included vaporized H2O2 as a Category A sterilizing agent. “These are methods that have a long history of safe and eff effective as demonstrated through multiple sources of information such as ample literature, clearances of 510(k)s or approvals of premarket approval (PMA) applications, and satisfactory QS inspection.”.*14
- Adaptation of processes and modifications to incorporate the expected alternative practices EMA prefers will temporarily reduce capacity and entail substantial capital/ operating costs.
- Using the less desirable non-isonon-isolator CRABS processes maintains a reliance on manual decontamination of critical environments, which is decidedly less effective than the use of an automated vapor phase H2O2 treatment.
* FDA considers decontamination a practice that embraces both sterilization and disinfection processes
Conclusion
The stated goals of the Annex 1 revision were the incorporation of enhanced knowledge and advances in technology. There’s a quotation. “Lead, follow, or get out of the way,” attributed to George Patton, that speaks to the subject of this article. In my opinion, the latest revision of EMA’s Annex 1 fails those objectives. It does not lead - its support for the best available technologies is minimal. It does not follow - there are many publications documenting the superiority of isolation technology. 4 It does not ‘get out of the way’ - there are constraints and obstacles imposed on isolators relative to less capable alternatives.
Current designs for aseptic filling present isolators as the ‘Best Available Technology’ for sterile product manufacture. The rapidly growing adoption of gloveless isolators for aseptic processing provides a level of asepsis that could only be dreamed of a decade ago. EMA’s approach in Annex 1 has the effect of promoting obsolescence and half measures over a substantially more promising future.
References
- EMA, Annex 1, Manufacture of Sterile Medicinal Products, 2022
- Morris, W. PDALetter, p. 30-31, July-August 2008
- Dorn, E. & Valerio, P., “ISPE Barrier Survey 2020”, Presentation at ISPE Aseptic Conference, Bethesda, March 2020.
- Agalloco, J., & DeSantis, P., “Technologies for Aseptic Filling: The Choice is Clear”, American Pharmaceutical Review, Vol. 27, No. 2, pp. 36-39, 2024.
- Whyte, W., Cleanroom Design, 2nd Edition, John Wiley & Sons, Chichester, 1999.
- B. Ljundvist, B., and Rheinmuller, B., “Modern Cleanroom Clothing Systems: People as a Contamination Source, PDA Journal of Pharmaceutical Science & Technology, 57 (2), pp. 114-12,5, 2003.
- 7. ISO 14644-7, Cleanrooms and Associated Controlled Environments — Part 7: Separative devices (clean air hoods, gloveboxes, isolators and minienvironments), 2004.
- Akers, J., Agalloco, J., Madsen, R., “What is Advanced Aseptic Processing?”, Pharmaceutical Manufacturing, Vol 4, No.2, pp 25-27, 2006.
- Bozenhardt, H., & Bozenhardt, E., “What, You Call That A RABS?” 7 (Real-Life) Aseptic Filling Blunders To Avoid, online at https://www. pharmaceuticalonline.com/doc/what-you-call-that-a-rabs-real-life aseptic-filling-blunders-to-avoid-0001 ,2019
- Kline, S., “Aseptic Processing Facility Design”, in Advanced Aseptic Processing Technology, edited by Agalloco, J. & Akers, J., Informa Healthcare, London, 2010.
- Hopkins Blog post Medicines and Healthcare Products Regulatory Agency. MHRA Inspectorate Blog: VHP (Vapour Hydrogen Peroxide) Fragility. https://mhrainspectorate. blog.gov.uk/2018/04/20/vhp vapour-hydrogen-peroxidefragility/, 2018.
- USP, Vapor Phase Sterilization, 2016
- Pierobon, C., “Challenges in Sterilizing Indirect Product Contact Surfaces – The Stopper Bowl Dilemma”, PDALetter, March 2025.
- FDA, Notification (510(k)) Submissions for Devices Labeled as Sterile Guidance for Industry and Food and Drug Administration Staff, 2024.
Subscribe to our e-Newsletters.
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!