Frank A. Chrzanowski, PhD- President, ExpertPharma, LLC
Abstract
The Standard Operating Procedures (SOPs) created by Artificial Intelligence (AI) using Microsoft CoPilot Software were compared to the Procedures provided in Previously Published Manuscripts to determine if AI assisted and improved the Procedures. The Previously Published Manuscripts reported Preformulation activities applied to resolve problems. Three problems were addressed: (a) Salt Selection for development of a Controlled Release Oral Product; (b) Improving the problematic stability of Cetiedil Citrate Injection and its Shelf-Life; and (c) Development of a Poorly Soluble Retinide, Fenretinide, for oral administration. The AI-generated SOPs provided extensive procedures, including some related to other departments such as Analytical and Regulatory. Comparisons of the Generated Procedures applicable to Preformulation and the specific problem were in excellent agreement with the Previously Published Manuscripts. There were differences in language and organization. AI demonstrated its ability to assist and improve the creation of SOPs, which is valuable in the absence of an SOP or in updating and improving an existing SOP.
Introduction
The ability of artificial intelligence (AI) to assist and improve procedures involved in the development of pharmaceuticals was evaluated by comparing a generated standard operating procedure (SOP) for a given preformulation problem to previously published manuscripts for the same problem. The author provided six published manuscripts detailing the resolution of preformulation problems in the areas of (a) salt selection for the development of a controlled-release oral product; (b) improving the problematic stability of cetiedil citrate injection and its shelf-life; and (c) development of a poorly soluble retinide, fenretinide, for oral administration.
Materials and Methods
Salt Form Selection
Salt form selection is performed at the earliest stage of development, after the first scale-up batch of the drug substance is used to create candidate forms. Optimal forms are identified rather than the best form because the best form would require more and longer testing. The procedure described can usually be completed within 1 – 2 weeks, facilitating further development.
The salt form selection was addressed in two publications: Preformulation Considerations for Controlled Release Dosage Forms: Part I, Selecting Candidates,1, and Part III. Candidate Form Selection Using Numerical Weighing and Scoring.2 The first manuscript describes the physical-chemical evaluation of eight forms of McN-5707, the free base, hydrochloride, sulfate, phosphate hydrate, fumarate, maleate and 2-napsylate salts in five fundamental areas, (a) organoleptics and equivalence factor CF; (b) crystallinity and melting behavior; (c) hygrodynamics, drying and hygroscopicity; (d) processability, filming, sticking on compression and rusting tooling, (e) critical solubilities, water, 0.1 N hydrochloric acid and pH 7.5 buffer. The optimal forms were identified in Part I by disqualifying any form for one or more seriously problematic properties. Part III applies two numerical analysis methods, Decision Analysis (DA) and Potential Problem Analysis (PPA), to identify optimal forms and compare DA, PPA, and Logical selection methods.
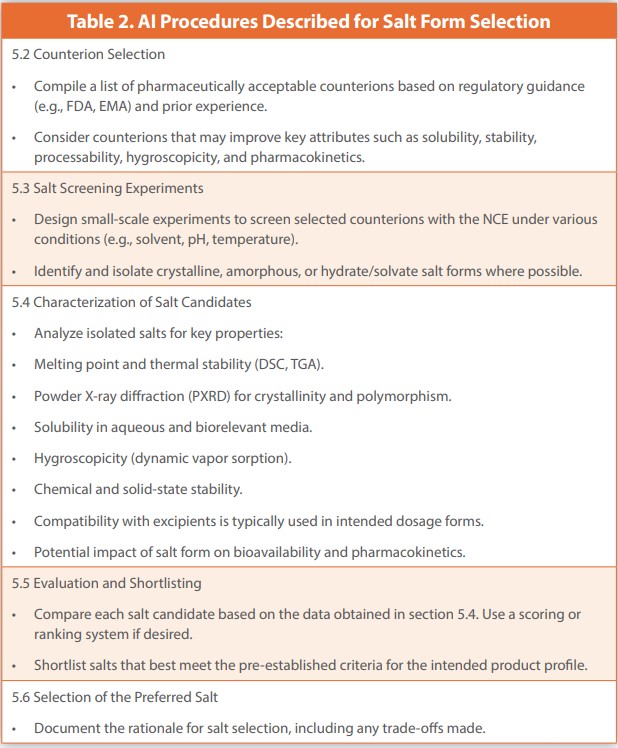
CoPilot was instructed to create a standard operating procedure for salt selection of a new chemical entity (NCE). It created a Standard Operating Procedure for Salt Selection of a New Chemical Entity (NCE) with a subtitle, Ensuring Optimal Formulation and Clinical Development. The term controlled release was not included in the AI instructions because procedures for immediate and controlled release are identical.
Cetiedil Citrate Injection Stability and Shelf-Life
The Cetiedil Citrate Injection Stability and Shelf-Life Problem was published as a case study. Cetiedil Citrate Injection contains Cetiedil Citrate, Sodium Chloride, and Water for Injection in a multiple-dose vial intended for single use. The MDVs are terminally sterilized with heat and pressure. The solution is unbuffered and has a pH in the range 3.3 to 3.8. The pH-stability profile shows that pH 3.3 to 3.8 is the pH of maximal stability. At the beginning of clinical and dosage form stability studies, it became evident that the solution was insufficiently stable at ambient temperature, 25˚C. A Preformulation Study was initiated that investigated the concentration, pH, buffers, and temperature effects as a means of improving the stability and providing a suitable shelf-life. Solution kinetic studies were performed using accelerating temperature conditions over the range pH 1 to 5 to obtain degradation rates (k). The degradation rates obtained at pH 3.4 and pH 5 were applied to an Arrhenius plot, and the extrapolated values of k at 25˚C and 8˚C were applied to the steps involved in Manufacture, Quality Control, Quality Assurance, Transport, and Storage to demonstrate the effects of temperature control on the stability and shelf-life of cetiedil injection. The data supported a shelf life of >4.2 years, <5% degradation.
CoPilot was requested to create a Standard Operating Procedure (SOP) for the Improved Stability of an Injectable NCE Solution. It created a Standard Operating Procedure (SOP) for Improved Stability of an Injectable NCE Solution with a subtitle Ensuring Optimal Quality, Safety, and Efficacy of New Chemical Entity (NCE) Injectable Formulations.
Poorly Soluble Retinide, Fenretinide
The development of Fenretinide is detailed in three publications: Development of a Quantitative PXRD Method for both polymorphs,4, Excipient Compatibility, 5, and Stability of Fenretinid Polymorphs,6
Fenretinide, N-(4-hydroxyphenyl) retinamide, is a compound intended for certain kinds of cancer. The development of Fenretinide began under the direction of the Southwest Research Institute (SWRI) and continued at the RW Johnson Pharmaceutical Research Institute (RWJPRI). Samples of Fenretinide drug substance from lots acquired from SWRI and from early RWJPRI scale-up lots tested by PXRD and DSC revealed five different polymorphic forms and one binary mixture. The components of the binary mixture were identified as Forms I and II. As sscale-upcontinued to support clinical trials, the batches of drug substance continued to be either Form I or II, or the binary mixture of Forms I and II. To study the two forms more closely, quantitative PXRD methods were created, and 100% standards of Form I and Form II were identified. Stability studies of the two Forms and the mixture using HPLC and Quantitative PXRD demonstrated that Form II is the least stable form, has no effect on the stability of Form II in the binary mixtures, and Form II converts to an amorphous form, which then degrades rapidly.
Dosage form development began with a solid oral tablet. An Excipient Compatibility Study comparing DSC and Simulated Wet Granulation Isothermal Stress (SWGIS) Method showed the DSC method was unreliable, and the predictions of the SWGIS Method were both reliable and verified after two years of storage at 25˚. The bioavailability of the initial tablet was only about 1%. As an alternative, softgel formulations of Fenretinide suspended in vegetable oils with and without a surfactant were investigated, as was particle size. Bioavailability was improved and shown to be dependent upon the oil, surfactant, and particle size.
CoPilot was instructed to create a Standard Operating Procedure (SOP)for the Development of a Poorly Soluble Retinide with Two Polymorphic Forms. It created a Standard Operating Procedure (SOP) for the Development of a Poorly Soluble Retinide with Two Polymorphic Forms with a subtitle, Comprehensive Guidelines for Research, Development, and Characterization.
Results
General
Each of the CoPilot-generated SOPs contained all the sections relevant to an SOP, such as Purpose, Scope, Responsibilities, Definitions, Procedures, Regulatory Considerations, Quality Control and Risk Management, Training, References, and Appendix. In the absence of a procedural SOP, an AI-created SOP would be an asset. However, in comparing the generated SOPs with published manuscripts, most of the comparison is with the Procedure. The Published Manuscript Procedure is presented in Table 1, and the AI-Generated Procedure is presented in Table 2 with the outline numbered text as it appeared in the AI-Generated SOP. Some instructions or aspects of the Created SOP Procedures were eliminated from the table because of irrelevance such Instructions for Dissolution Testing for a compound such as Fenretinide which has almost zero solubility in aqueous media and pharmaceutical solvents; storage of an MDV in temperature-humidity conditions; directions normally assigned to other departments such as the development of a Stability Indicating Analytical Method and creation of the salts.

Salt Form Selection
The instructions from the Published Manuscript and Generated SOP are presented in Table 1 and Table 2, respectively. The two procedures are almost identical, except for the differences in language and more extensive instructions in the Generated Procedures, such as the use of Dynamic Vapor Sorption (DVS) and inclusion of pharmacokinetics. The availability of a DVS in place of controlled RH chambers is of minor concern. Pharmacokinetics would extend the testing appreciably and require more extensive resources, including additional amounts of each salt. The procedure is intended to quickly evaluate the salts using predominantly physical test methods and relying on simple UV spectrometric analyses in the solubility testing.
Several instructions provided by the AI-generated SOP are normative in-house practices that were not included in the published article. They include preparative activities such as a review of the chemical structure of the NCE, potential counterions obtained from a list of pharmaceutically acceptable counterions. Consideration to crystalline, amorphous, and hydrate/solvate forms where possible. (The Appendix lists hydrochloric, sulfuric, maleic, citric, tartaric, and methane sulfonic as acceptable counterions for a base.)Small-scale experiments are suggested to evaluate the PC properties of the salts. Consider counter ions that may improve key attributes such as solubility, stability, hygroscopicity, processability hygroscopicity and pharmacokinetics. Physical and chemical stabilities at ambient and higher temperatures and relative humidities, and evaluation in solution and slurry conditions. Prioritize counter ions with a history of safe use. Final selection should include a process for the identification of optimal forms. All of which were performed as an internal corporate normative activity but omitted in the Published Manuscripts 1,2.
Cetiedil Citrate
The instructions from the Published Manuscript and Generated SOP are presented in Table 3 and Table 4, respectively. The obvious differences between Table 3 and Table 4 are that the instructions are similar, but more extensive in Table 4, and there are instructions for environmental contro,l which were applied to create longevity tables that defined the concentrations remaining after each step in manufacturing, , QC and QA, transportat, ion and storage using the degradation rate constants obtained from the stability study. The longevity table, which is not shown, demonstrated that the use of a Cold Place (2˚ to 8˚ C) for storage and transport had a significant effect on stability and shelf-life, with <5% degradation in > 4.2 years.


Fenretinide
The instructions from the Published Manuscript and Generated SOP are presented in Table 5 and Table 6, respectively. Again, the instructions in both tables are comparable, but differ in expression and sometimes in the extent of testing. Preformulation partnered with Chemical Development to develop a recovery process that always produced the intended forms I. However, each trial was not always successful, such that there were always available Form 2 and the binary mixture. PXRD and DSC gave early indications that Form I was more stable than Form II and would be the lead polymorph. The addition of HPLC to Quantitative PXRD showed that Form II converted to an amorphous form with little degradation, but the amorphous form was the most unstable.
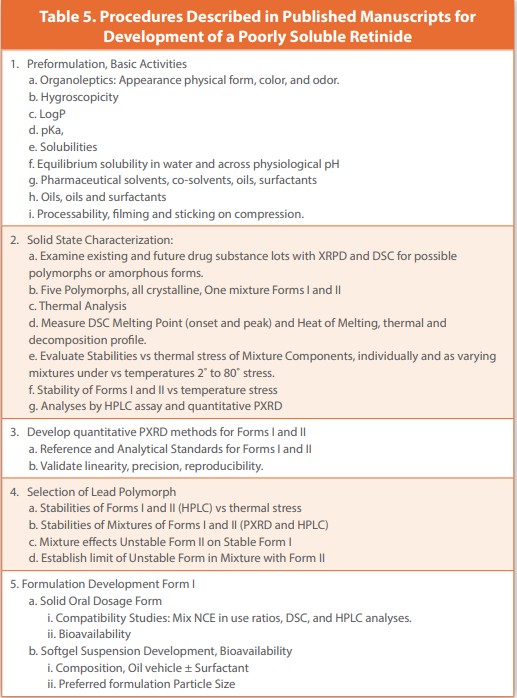
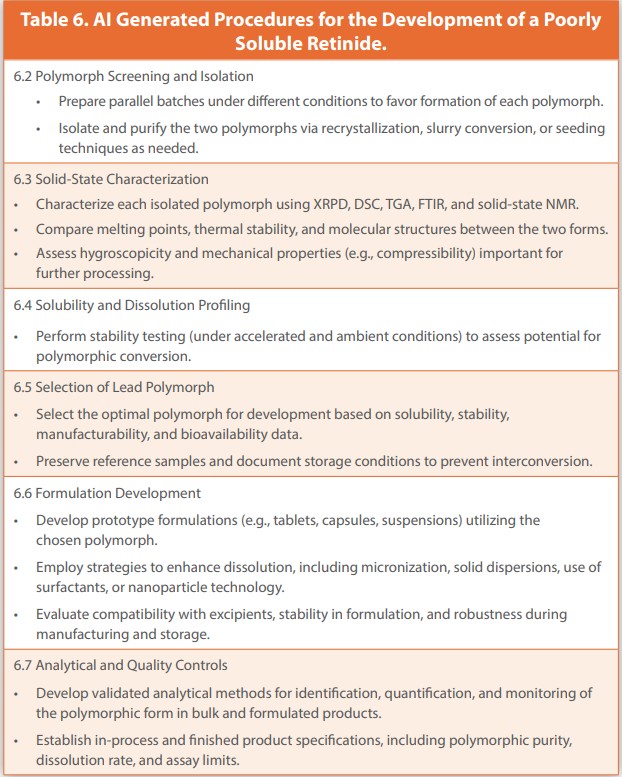
An excipient Compatibility study of solid dosage form excipients showed that there were no incompatibilities when the test was done with a SWGIS Method; however, that formulation had near-zero bioavailability. Softgel formulations were the alternative applied. Stability was not an issue, and bioavailability was improved appreciably and shown to be dependent on the soft gel components and Fenretinide particle size.
Conclusion
The Microsoft Pilot AI-created SOPs are in very close agreement with procedures created to resolve preformulation problems. The differences observed were in language and depth of experimentation, which is favorable. A procedure can be modified, shortened, and even extended, given that it is stated. Instructional assigned procedures were also included for other departments and resources such as Chemical Development, Chemical Analytics, Dissolution, and Regulatory, which is a positive. In-house practices that are not defined in an SOP also appeared in the AI SOP, another positive.
The AI-generated SOPs included procedures described in the Published Manuscripts. In the absence of an existing Operating Procedure or where operating procedures are being updated, the experiences of this comparison study indicate that Microsoft CoPilot and AI could be used to create new SOPs and improve existing SOPs.
References
- Chrzanowski FA. Preformulation Considerations for Controlled Release Dosage Forms. Part I: Selecting Candidates. AAPS PharmSciTech 2008 June; 9(2):635-8. Epub 2008 Mar 28. doi: 10.1208/s12249-008-9068-2.
- Chrzanowski FA. Preformulation Considerations for Controlled Release Dosage Forms. Part III Candidate Form Selection Using Numerical Weighting and Scoring. AAPS PharmSciTech 2008 June; 9(2): 646–650. Epub 2008 May 24. doi: 10.1208/s12249-008-9069-1.
- Chrzanowski FA. Cetiedil Citrate Injection, Resolve Instability: Case Study. J Clin Pharm 2020;4(1):1016-1023.
- Chrzanowski FA, Fegely BJ, Sisco WR, Newton MP. Analysis of N-(4-hydroxyphenyl) retinamide Polymorphic Forms by X-ray Powder Diffraction. J Pharm Sci 1984;73:1448- 1450.
- Chrzanowski FA, Ulissi LA, Fegely BJ, Newman AC. Preformulation Excipient Compatibility Testing. Application of a Differential Scanning Calorimetric Method versus a Wet Granulation Simulating, Isothermal Stress Method. Drug Dev and Ind Pharm 1986;12:783- 800.
- Walking WD, Sisco WR, Newton MP, Fegely BJ, Chrzanowski FA. Stability of Fenretinide Polymorphs. Acta Pharm Technol 1986;32:10-12.
Acknowledgements
The author acknowledges that the previously published manuscripts represent preformulation activities while employed by McNeil Pharmaceutical, Spring House, PA 19076. The co-authors and colleagues in the six manuscripts are: Walkling WD, Fegely BJ, Ulissi LA, Newman AC, Sisco WR, Newton MP, Egan RS, Ko CY, Paragamian V, Mills JE, Plampin JN, Kotwal P, Janicki C.
The two Salt Form Publications represented work presented at the 41st Annual Pharmaceutical Technologies Arden Conference, January 2006, West Point, NY.
Funding Statement
The author is currently semi-retired from employment with McNeil Pharmaceutical and Johnson & Johnson Companies. This manuscript preparation was unfunded. However, all of the work reported occurred as an employee of McNeil Pharmaceutical, a Johnson & Johnson Family Company.
Conflict of Interest
The author has no conflicts of interest related to this publication and the previously cited publications. Orchid ID: 0000-09003-4391-7989 Frank A Chrzanowski, FA Chrzanowski, Francis A. Chrzanowski
Author Biography
Frank Chrzanowski is a semi-retired Pharmaceutical Scientist with over 40 years’ experience in the development of Pharmaceuticals. His experience includes 20 years supporting dosage form development, heading the Preformulation Laboratories at McNeil Pharmaceutical and the RW Johnson Pharmaceutical Research Institute, and over 20 years as a Consultant and Expert/Expert Witness in patent litigation cases in the US and Canada. He has held Assistant Professor positions at the Massachusetts College of Pharmacy and Northeastern University, as well as an Adjacent Assistant Professor. Prof Position at the Univ of Cincinnati.
He was also responsible for the development of liquid oral and parenteral dosage forms, and extemporaneous dosage forms intended for bioavailability, pharmacokinetic, and drug safety studies in humans and animals.
He has served as an Expert/Expert Witness in US and Canadian Notice pharmaceutical patent litigation cases. He has been an invited speaker for AAPS programs and College Seminars.
Dr. Chrzanowski has a BS (Pharmacy), MS, and PhD (Pharmaceutics) degrees, all from the Philadelphia College of Pharmacy and Science (Pharmaceutics), which is now a part of St Joseph’s University (Philadelphia).
Subscribe to our e-Newsletters.
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!