Introduction
Before SFC was coupled to mass spectrometers, systems used both UV and flame ionization detectors (FID). This may not seem intuitive, however, when one considers that supercritical fluid chromatography (SFC) is essentially a hybrid of gas chromatography and liquid chromatography (supercritical CO2 is composed of gaseous carbon dioxide molecules pressed as closely together as they can be, without forming a liquid), then the use of both these detectors makes sense.
Sources
The most prevalent SFC-MS sources are the atmospheric pressure ionization (API) sources, used in tandem with packed columns. Most of the columns commonly used for normal phase chromatography, both chiral and achiral, can be used with SFC [1, 2]. Integrating SFC and a mass analyzer is fairly straightforward and is easier than LC-MS, as CO2 is highly volatile, which makes it easier to convert effluents to the gas phase during the ionization process. The earlier SFC-MS ionization sources followed the evolution of the ionization sources for LC-MS and GC-MS and as these sourcess continue to improve, they too are applied to SFC [3, 4].
The most popular ionization sources for SFC-MS are atmospheric pressure chemical ionization (APCI) and electrospray ionization (ESI) as they allow direct infusion of the chromatographic effluents into a mass analyzer. Both ESI and APCI are used in the pharmaceutical industry, with ESI being more prevalent for LC-MS applications. The use of these two sources in LC-MS is based on ionizability in solution, molecular weight, volatility, polarity and flow rate. It is possible to use these sources without further modification for SFC-MS applications; [5] however some modifications may be required to enhance ionization and achieve a stable baseline [6].
Atmospheric Pressure Chemical Ionization (APCI)
The APCI is the most common source currently used in SFC-MS. It is easily adaptable to SFC systems and can handle the high flow rates of a typical SFC method (2 to 5 mL/min). Since it is coupled with a highly volatile mobile phase, it does not suffer from reduced ionization efficiency as may be experienced in LC-MS at higher flow rates. The source is durable and easily cleaned. It is a mass dependent source, which means the greater the amount sample, the better the sensitivity. It is less aff ected by ion pairing agents, than its electrospray counterpart, which also translates into being less aff ected by ion suppression than ESI. The key parameter in APCI is the probe temperature, where one has to fi nd the temperature where there is maximum sensitivity coupled with minimum decomposition.
The mechanism of ionization in APCI is vaporization > solvent(X-OH) ionization > charge transfer to analyte. The most common ionization reactions, starting from atmospheric nitrogen, in the APCI source are: [7]

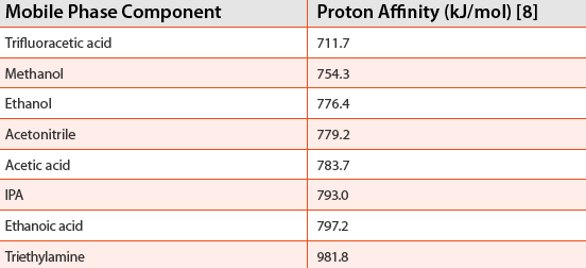
Since carbon dioxide does not have a proton to donate, use of APCI usually requires the addition of hydrogen donors (X-OH). The proton transfer reaction occurs when the proton affinity of the analyte molecules is greater than that of the reagent gas/mobile phase component.
As an example, from Table 1, acetonitrile could transfer a proton to triethylamine, but not to methanol.
Table 1. Proton affinities of some SFC additives.
Electrospray Ionization (ESI)
Electrospray ionization occurs as the solvent is nebulized, followed by evaporation to leave ionized particles in the gas phase. A high electrical field (~3 kV) is applied between the needle and a nearby counter electrode, during nebulization. The resulting charged droplets shrink via evaporation, leaving small highly charged particles that are propelled towards the mass analyzer. ESI, in contrast to APCI, produces fewer ion fragments for most polar compounds [9]. However, ESI works best with ionizable buffers, which is why it is a favourite for reverse phase LC-MS and why it is less popular with SFC, as the mobile phases in SFC tend to be more non-aqueous in nature.
Atmospheric Pressure Photoionization (APPI)
Atmospheric pressure photoionization is used less than APCI or ESI but can be useful, especially with polyaromatic hydrocarbons or steroids. APPI uses a lamp (usually krypton) that has energy of 10.6 eV. Ionization occurs when the ionization potential of the lamp exceeds that of the analyte. Solvents that have ionization potentials (IP) that are higher than the lamp absorb the energy and little to no ionization occurs as the energy is dissipated in the solvent.
"The workhorse mass analyzer for SFC-MS in the pharmaceutical industry is the single quadrupole mass spectrometer. This type of mass spectrometer is ubiquitous in research and development laboratories, easily controlled by existing software and is easily adaptable to sources and interfaces."
Ionization occurs when a photon interacts with an analyte (Manalyte) to create a radical ion, which can then be protonated by protic solvent molecules:
Depending on the protic solvent it is possible to have quantities of both Manalyte +∙ and [Manalyte +H]+. If there is no source of protons, dopants (D) with low ionization potentials may be added, post separation, presource to enable protonation of the analyte.
Examples of dopants include acetone with an IP of 9.72 eV and toluene with an IP of 8.82 eV [10]. It has been suggested that APPI is less susceptible to sample matrix effects [11], making it attractive for the analysis of drugs or metabolites in tissues, blood or plasma and works well for very non-polar molecules [12]. APPI can be more sensitive than APCI or ESI, as the common solvents used in SFC do not ionize well under APPI conditions. This leads to lower background ionization and noise.
Source and Mass Analyzer Interfaces
The ionization source is only a part of the setup in an SFC-MS system. How the sources are coupled to the mass analyzers is also important. There are several options, some of which are more universal or user friendly and others that are more sensitive. This is more straightforward in LC-MS, whereas the phase behavior of the CO2 makes these interfaces more challenging. As the SFC mobile phase elutes from the column, the pressure on the gas is released, the density decreases, the CO2 can boil off and any dissolved components can precipitate. It is therefore necessary for the interface that links the ionization source to the mass spectrometer to control or counteract these problems.
- Interfaces used in SFC-MS include [6]: Splitting the flow prior to the back pressure regulator (BPR) ĔĔ This is the configuration that is most often found with instrument manufacturers/ vendors. This configuration splits the flow after the column, such that most of the column flow goes to the UV detector and only a portion goes to the MS (similar to LC-MS) setups. The BPR is placed in-line after the UV detector. This approach is easily controlled by the instrument software and generally maintains good chromatographic fidelity [13]. There are however, a few disadvantages, one being only a portion of the effluent is diverted to the source, which can affect sensitivity when using mass dependent ionization sources.
- Injecting the total flow from SFC into the MS using a pressure regulating fluid interface ĔĔ To counteract variability due to splitting the flow above, a liquid that is miscible with CO2 that is teed into the column effluent by a zero dead volume connector can be used. This miscible liquid works as the pressure regulator and all the effluent is then pumped into the ionization source
- Injecting the total fl ow from SFC into the MS using a BPR Ĕ The total eluent fl ow can be introduced into the mass spectrometer using a mechanical back pressure regulator (BPR). Since commercial BPRs are not necessarily able to maintain good chromatographic fi delity, for this approach to work all large volume components must be removed from the fl ow path. The main advantages of this setup are that the total effl uent fl ow goes to the mass spectrometer and that the instrumental parameters are more easily controlled by the system’s software. As long as the dead volume in the BPR and transfer lines is minimized the chromatographic separations remain intact.
- Injecting the total fl ow from SFC into the MS using passive BPR Ĕ The simplest interface to the mass spectrometer is one in which the total effl uent is directed towards the mass spectrometer; however the back pressure is controlled with a single piece of restrictor tubing.
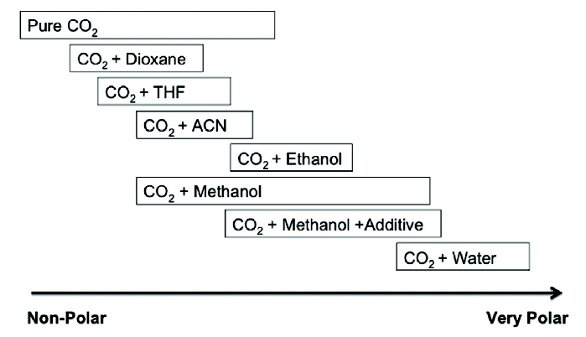
Figure 1. Schematic showing polarity ranges of mobile phase combinations in SFC-MS.
With all of these interfaces, there are still the underlying issues around how easy it is to validate a method with the above confi gurations. This is of particular concern in the pharmaceutical industry as some companies outsource their analytical work and with manufacturing now happening around the globe, transferable, robust and validatable methods are a must. The upswing in interest in SFC and new instrumentation on the market, coupled with careful control of the instrumental parameters, has made it easier to validate and transfer SFC methods.
Mass analyzers
All the above mentioned sources and interfaces need next to be connected to a mass analyzer. Similar to LC-MS, the choice of mass analyzer depends on the application.
The workhorse mass analyzer for SFC-MS in the pharmaceutical industry is the single quadrupole mass spectrometer. This type of mass spectrometer is ubiquitous in research and development laboratories easily controlled by existing software and is easily adaptable to the sources and interfaces mentioned above. Single quadrupole mass spectrometers are particularly useful for both achiral [14] and chiral [15] high throughput screening.
One of the more prevalent mass analyzers used in the analysis of trace components in complicated matrices is the triple quadrupole mass analyzer. The quadupoles fi lter out any interference or matrix eff ects, as interfering components would have to contain the same initial ion fi ltered by the fi rst quadrupole and the same daughter product selected by the third quadrupole to cause any interference.
Time-of-flight (TOF) mass spectrometers couple well with high-speed separation techniques, like SFC, due to a high rate of data acquisition. Other examples of mass analyzers combined with SFC include: Q-TOF [16], Fourier Transform [17], ion trap [18], magnetic sector [19] and double focusing [20, 21] mass spectrometers. However, with ultra high pressure chromatography (UHPLC) maturing, new single quadrouple and other mass analyzers capable of higher scan speeds are being developed to be coupled to the ultra high pressure systems. In the future, these newer systems could also be coupled to SFC systems.
Types of analyses
SFC-MS analyses can be used for a wide range of analyses ranging from normal phase type analyses to chromatography that more emulate reverse phase chromatography by using small amounts of water as an additive. Figure 1 shows the mobile phase and polarity range for supercritical fluid chromatography.
Chiral SFC-MS
Chiral SFC has many advantages over normal phase liquid chiral chromatography. The first advantage is its speed; due to the low viscosity of CO2, SFC can be run at much faster flow rates than normal phase HPLC, even when employing columns of the same dimensions. This leads to faster equilibration, even when changing solvents (again this emulates gas chromatography, more than liquid chromatography). The second advantage is that SFC chromatography is easily scaled up to the preparatory level, and the recovery of enantiomers is much simpler as the amount of solvent that needs to be removed post SFC separations is minimal by comparison. This is especially useful during the early phases of drug discovery. Unlike normal phase chromatography, however, where the most common modifier is isopropyl alcohol (IPA), the preferred starting point for method development would be with methanol as SFC-MS modifi er. Due to its lower surface tension, it gives better ionization efficiency and therefore sensitivity in the mass spectrometer over other alcohols, such as ethanol and IPA [13]. Many pharmaceutical investigators are looking at using SFC-MS for screening or automating chiral separations.
Achiral SFC-MS
Achiral SFC-MS can cover a very broad range of compounds. There are examples of using SFC for the fast analyses and the detection of codeine in urine and other alkaloids, as well as phenylbutazone and its metabolites, also from urine [22]. As the SFC technology and sensitivity increase, there has been an increase in achiral applications including in-process testing, due to the speed (keeping up with UHPLC methods) and reduced waste, in comparison to traditional normal phase methods.
Prep SFC-MS
Mass-directed fraction collecting has been used with prep SFC to separate the enantiomers of several pharmaceutically relevant compounds including: warfarin, metoprolol, promethazine, cortisterone, tolbutamide, coumarin, atenolol, imipramine, alprenolol, erythromycin and verapamil [23]; underscoring that prep SFC-MS can be used instead of prep SFC-UV for the isolation and purification of enantiomers. After resolving some of the earlier problems, such as proper integration of the software, optimizing the interface between the mass spectrometer and the SFC and loss of sample at the fraction collector due to aerosol formation, there are successful examples in the pharmaceutical industry of regular use of mass-directed for the purification of their library compounds [24].
Due to the need for chirally and achirally pure active pharmaceutical ingredients, there has been more interest in prep SFC-MS gradient systems. Evaluation of such systems currently shows that high recoveries are possible and that the instrumentation produces good gradients, reasonable flow rates and is capable of mass-directed fractionation [25]. Prep SFC-MS systems have also been applied to achiral separations of active pharmaceutical ingredients (API). The approach is desirable for the same reasons as using prep SFC-MS for chiral work, including different selectivities, highly concentrated fractions due the easy removal of CO2 and high recovery rates [26].
Some industry investigators have combined both achiral and chiral purifications in their 2D SFC/SFC/MS system, which produces two-step purifications in one process. The first dimension consists of an achiral separation to remove impurities from the chiral compounds and the second dimension completes the chiral separations. The evolution of such 2D SFC/SFC/MS systems could prove to be the most efficient way to purify chiral compounds [27].
Structure Elucidation
Since in most cases, the bulk of the mobile phase in SFC-MS is CO2 and only small volumes of additives are used, the use of deuterated solvents (i.e. d4-MeOD) becomes a very accessible, inexpensive way of determining the location of exchangeable protons or studying H-D exchange reactions. This is an area of SFC-MS that is expected to develop as more analytical SFC-MS systems come online.
Conclusion
Traditionally, SFC-UV (and, to a certain extent, SFC-MS) has suffered from a lack of sensitivity. Many new advancements in the past few years have increased the sensitivity and stabilized the baseline in SFCMS. Overall, the future applications of SFC-MS look bright.
Author Biography
Dr. Laila Kott is currently at Millennium (The Takeda Oncology Company) in the Analytical Development Small Molecule department where she is a group lead developing methods for early and late stage projects. She received her Ph.D. from the University of Massachusetts and has publications on many analytical topics.
References
- Zhao, Y., Sandra, P., Woo, G., Thomas, S., Gahm, K. and Semin, D. Pharm. Discovery, (2005) 5, 30-41.
- Wang, Z., Li, S. Jonca, M., Lambros, T., Ferguson, S. Goodnow, R. and Ho, C. Biomed. Chromatogr., (2006) 20, 1206-1215.
- Zaugg, S.D., Deluc, S.J., Holzer, G.U. and Voorhees, K.J. J. High Resolut. Chromatogr. Chromatogr. Commun., (1987) 10, 100-101.
- Arpino, P.J., Dillettato, D., Nguyen, K. and Bruchet, A.J. J. High Resolut. Chromatogr., (1990) 13, 5-12.
- Morgan, D.G., Harbol, K.L. and Kitrinos, P.N., Jr. J. Chromatogr. A, (1998) 800, 39-49.
- Pinkston, J.D. Eur. J. Mass. Spectrom., (2005) 11, 189-197.
- Niessen, W. M.A. Liquid Chomatography—Mass Spectrometry, 3rd Ed., 2006, Taylor & Francis, USA.
- Hunter, E. P. and Lias, S. G. J. Phys. Chem. Ref. Data, (1998) 27, 413-656.
- Li, F. and Hsieh, Y. J. Sep Sci., (2008) 31, 1231-1237.
- Brundle, C. R., Robin, M. B., Kuebler, N. A. Basch, H. J. Am. Chem. Soc., (1972) 94, 1454-1465.
- Hsieh, Y., Merkle, K., Wang, G., Brissen, J.-M. and Korfmacher, W. A. Anal. Chem., (2003) 75, 3122-3127
- Bolaños, B., Grieg, M., Ventura, M., Farrell, W., Aurigemma, C. A., Li. H., Quezner, T. L., Tivel, K., Bylund, J. M. R., Tran, P., Pham, C. and Phillipson, D. Int. J. Mass. Spectrom., (2004) 238, 85-97.
- Zhao, Y., Woo, G., Thomas, S., Semin, D., and Sandra, P. J. Chromatogr. A, (2003) 1003, 157-166.
- Ventura, M.C., Farrell, W.P., Aurigemma, C.M. and Grieg, M.J. Anal. Chem., (1999) 71, 2410-2416.
- Zeng, L., Xu, R., Laskar, D. B. and Kassel, D. B. J. Chrom. A, (2007) 1169, 193-204.
- Garzotti, M., Rovatti, L. and Hamdan, M. Rapid Comm. Mass. Spec., (2001) 15, 1187-1190.
- Lee, E. D., Henion, J. D., Cody, R. B. and Kinsinger, J. A. Anal. Chem., (1987) 59, 1309-1312.
- 46 Todd, J. F. J., Mylchreest, I. C., Berry, A. J., Games, D. E. and Smith, R. D. Rapid Comm. Mass. Spec., (1988) 2, 55-58.
- Kalinoski, H. T., Udseth, H. R., Chess, E. K. and Smith, R. D. J. Chrom., (1987) 394, 3-14.
- Ekeberg, D. and Jablonska-Jentoft, A. M. J. Am. Soc. Mass Spectrom., (2007) 18, 2173-2179.
- Reinhold, V. N., Sheeley, D. M., Kuei, J. and Her, G.-R. Anal. Chem., (1988) 60, 2719-2722.
- Crowther, J. B. and Henion, J. D. Anal. Chem., (1985) 57, 2711-2716.
- Wang, T., Barber, M., Hardt, I., and Kassel, D.B., Rapid Comm. Mass. Spec., (2001) 15, 2067- 2075.
- Zhang, X., Towle, M. H., Felice, C. E., Flament, J. H. and Goetizinger, W. K. J. Comb. Chem., (2006) 8, 705-714.
- Ebinger, K. Weller, H. N., Kiplinger, J. and Lefebvre, P. JALA, (2011) 16, 241-249.
- Van Anda, J. Am. Pharm. Review, (2010) 13, 111-115.
- Zeng, L., Xu, R., Zhang, Y. and Kassel, D. B. J. Chromatogr. A, (2011) 1218, 3080-3088.