Quality by Design (QbD) is a concept first developed by the quality pioneer Joseph M. Juran.1 Juran believed that quality should be designed into a product and that most quality crises and problems relate to how a product was designed in the first place. A high-quality drug product is a product free of contamination and reliably delivers the therapeutic benefit promised on the label to the consumer.2 The United States Food and Drug Administration (US-FDA) encourages risk-based approaches and the adoption of QbD principles in drug product development, manufacturing, and regulation. FDA’s emphasis on QbD began with the recognition that increased testing does not necessarily improve product quality. Quality must be built into the product.5
The concept of biopharmaceutical Quality by Design and its objectives include the following elements:
- Quality Target Product Profile (QTPP)
- » A quality target product profile that identifies the critical quality attributes of the drug product.
- Critical Quality Attributes (CQA)
- » A physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality,6 Consider all DP (Drug Products) quality attributes; physical attributes, identification, assay, content uniformity, dissolution and drug release, degradation products, residual solvents, moisture, microbial limits, etc.
- Identify a CQA based on the severity of harm to a patient (safety and efficacy) resulting from failure to meet that quality attribute. » Identified before taking into account risk control.
- Does not change as a result of risk management.
- Critical Process Parameter (CPP)
- A process parameter whose variability has an impact on a CQA and therefore should be monitored or controlled to ensure the process produces the desired quality.6
- Critical Material Attribute (CMA)
- A physical, chemical, biological, or microbiological property or characteristic of an input material that should be within an appropriate limit, range, or distribution to ensure the desired quality of output material. CMA is not defined in ICH guidances.
As the biopharmaceutical industry moves toward the implementation of biopharmaceutical QbD, a common terminology, understanding of concepts and expectations are necessary. This understanding will facilitate better communication between those involved in risk-based drug development, manufacturing, and drug application review, Figure 1.
Over the years, biopharmaceutical QbD has evolved with the issuance of ICH Q8 (R2), (Biopharmaceutical Development), ICH Q9 (Quality Risk Management), and ICH Q10 (Biopharmaceutical Quality System).3–5 In addition, the ICH Q1WG on Q8, Q9, and Q10 Questions and Answers; the ICH Q8/Q9/Q10 Points to Consider document; and ICH Q11 (Development and Manufacture of Drug Substance) have been issued, as have the conclusions of FDA-EMA’s parallel assessment of Quality-By-Design elements of marketing applications.6–9 These documents provide high-level directions concerning the scope and definition of QbD as it applies to the biopharmaceutical industry, Figure 2.
Many implementation details are not discussed in this guidance or document. There is confusion among industry scientists, academicians, and regulators despite recent publications.10–13
Quality by Testing Versus Quality by Design
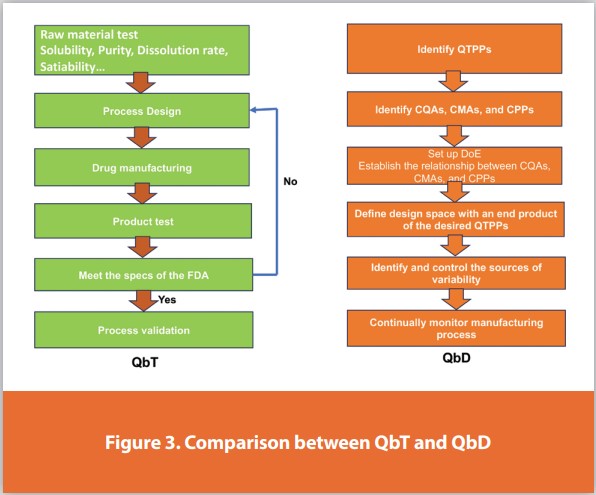
The biopharmaceutical sector is concerned with product efficacy, safety, and quality. FDA decided to implement various Designs of Experiments (DoE) in the biopharmaceutical industries. Quality by Design helps in the development of high-quality products and guides to manage product quality across its entire life cycle. This method quickly gained popularity worldwide due to its many benefits and use of numerous high-quality statistical tools. QbD is a meticulous, proactive, risk-based strategy for pharmaceutical development that commences with predetermined goals and emphasizes product and process understanding and process control based on reliable research. The QTPP, knowledge of product and process design, scale-up, control strategy, and constant improvement are considered crucial components of pharmaceutical QbD. After approval, during the product lifecycle management process, the competency of the product and the process is evaluated and continuously enhanced, Figure 3.
Quality by Design (QbD) is a systematic approach to biopharmaceutical development that begins with predefined objectives. It emphasizes product and process understanding and process control, based on sound science and quality risk management, Figure 4.
- Specification acceptance criteria are based on performance.
- Testing may not be necessary to release batches.
Quality by Testing (QbT) is the process developers use to verify and assure the quality of a product or service before releasing it to customers. QA testing doesn’t involve creating the product, but rather examining the quality of the end product and the outcomes it can achieve:
- Specification acceptance criteria are based on one or more batch data (process capability).
- Testing must be made to release batches.
Concept of Process Capability
- First introduced in the Statistical Quality Control Handbook by the Western Electric Company (1956). “Process capability” is defined as “the natural or undisturbed performance after extraneous influences are eliminated. This is determined by plotting data on a control chart.”
- ISO, AIAG, ASQ, and ASTM,29 published their guideline or manual on process capability index calculation.
Biopharmaceutical Quality by Design Objectives
Biopharmaceutical QbD is a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and control based on sound science and quality risk management.3 The goals of biopharmaceutical QbD may include the following:
- Achieve meaningful product quality specifications that are based on clinical performance.
- Increase process capability and reduce product variability and defects by enhancing product and process design, understanding, and control.
- Increase product development and manufacturing efficiencies.
- Enhance root cause analysis and post-approval change management.
Under QbD, these goals can often be achieved by linking product quality to the desired clinical performance and then designing a robust formulation and manufacturing process to consistently deliver the desired product quality, Figure 5.
Since the initiation of biopharmaceutical QbD, the FDA has made significant progress in achieving the first objective: performance-based quality specifications. Some examples of FDA policies include tablet scoring and bead sizes in capsules labeled for sprinkle products.14,15 The recent FDA discussions on the assayed potency limits for narrow therapeutic index drugs and physical attributes of generic drug products reflect this trend.16 Nonetheless, it should be recognized that ICH documents3-9 did not explicitly acknowledge clinical performance-based specifications as a QbD goal, although this was recognized in a recent scientific paper.10
The second objective of biopharmaceutical QbD is to increase process capability and reduce product variability that often leads to product defects, rejections, and recalls. Achieving this objective requires robustly designed sprinkle products. In addition, an improved product and process understanding can facilitate the identification and control of factors influencing the drug product quality. After regulatory approval, efforts should continue to improve the process to reduce product variability, defects, rejections, and recalls.
QbD uses a systematic approach to product design and development. As such, it enhances development capability, speed, and formulation design. Furthermore, it transfers resources from a downstream corrective mode to an upstream proactive mode. It enhances the manufacturer’s ability to identify the root causes of manufacturing failures. Increasing product development and manufacturing efficiencies is the third objective of biopharmaceutical QbD, Figure 6.

The final objective of QbD is to enhance root cause analysis and post-approval change management. Without good product and process understanding, the ability to efficiently scale up and conduct root cause analysis is limited and requires the generation of additional data sets on the proposed larger scale. FDA’s change Guidances17,18 provides a framework for post-approval changes. Recently, the FDA issued guidance intended to reduce the regulatory filing requirements for specific low-risk chemistry, manufacturing, and control (CMC) post-approval manufacturing changes.19
A specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria, which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a drug substance or drug product should conform to be considered acceptable for its intended use. “Conformance to specifications” means that the drug substance and/or drug product, when tested according to the listed analytical procedures, will meet the listed acceptance criteria. Specifications are critical quality standards that are proposed and justified by the manufacturer and approved by regulatory authorities as conditions of approval.31,32
The quality of drug substances and drug products is determined by their design, development, in-process controls, GMP controls, process validation, and specifications applied to them throughout development and manufacture. ICH Q6A addresses specifications, i.e., those tests, procedures, and acceptance criteria which play a major role in assuring the quality of the new drug substance and new drug product at release and during shelf life. Specifications are an important component of quality assurance but are not its only component. All of the considerations listed above are necessary to ensure consistent production of drug substances and drug products of high quality.31,32
Elements of Biopharmaceutical Quality by Design
In a biopharmaceutical QbD approach to product development, an applicant identifies characteristics that are critical to quality from the patient’s perspective, translates them into the drug product critical quality attributes (CQAs), and establishes the relationship between formulation/manufacturing variables and CQAs to consistently deliver a drug product with such CQAs to the patient. QbD consists of the following elements:
- A quality target product profile (QTPP) that identifies the critical quality attributes (CQAs) of the drug product
- Product design and understanding including the identification of critical material attributes (CMAs)
- Process design and understanding including the identification of critical process parameters (CPPs) and a thorough understanding of scale-up principles, linking CMAs and CPPs to CQAs.
- A control strategy that includes specifications for the drug substance(s), excipient(s), and drug product as well as controls for each step of the manufacturing process
- Process capability and continual improvement
Biological Activity
Assessment of the biological properties constitutes an equally essential step in establishing a complete characterization profile. An important property is the biological activity that describes the specific ability or capacity of a product to achieve a defined biological effect. A valid biological assay to measure the biological activity should be provided by the manufacturer. Examples of procedures used to measure biological activity include;
- Animal-based biological assays, which measure an organism’s biological response to the product
- Cell culture-based biological assays, which measure biochemical or physiological responses at the cellular level
- Biochemical assays, measure biological activities such as enzymatic reaction rates or biological responses induced by immunological interactions Other procedures such as ligand and receptor binding assays, may be acceptable.32
Quality Target Product Profile that Identifies the Critical Quality Attributes of the Drug Product
QTPP is a prospective summary of the quality characteristics of a drug product that ideally will be achieved to ensure the desired quality, taking into account the safety and efficacy of the drug product. QTPP forms the basis of design for the development of the product. Considerations for inclusion in the QTPP could include the following:3
- Intended use in a clinical setting, route of administration, dosage form, and delivery system(s)
- Dosage strength(s)
- Container closure system
- Therapeutic moiety release or delivery and attributes affecting pharmacokinetic characteristics appropriate to the drug product dosage form being developed
- Drug product quality criteria (e.g., sterility, purity, stability, and drug release) appropriate for the intended marketed product
Identification of CQAs of the drug product is the next step in drug product development. A CQA is a physical, chemical, biological, or microbiological property or characteristic of an output material including a finished drug product that should be within an appropriate limit, range, or distribution to ensure the desired product quality.3 The quality attributes of a drug product may include identity, assay, content uniformity, degradation products, residual solvents, drug release or dissolution, moisture content, microbial limits, and physical attributes such as color, shape, size, odor score configuration, and friability. These attributes can be critical or not critical. The criticality of an attribute is primarily based on the severity of harm to the patient should the product fall outside the acceptable range for that attribute. The probability of occurrence, detectability, or controllability does not impact the criticality of an attribute.
It seems obvious that a new product should be adequately defined before any development work commences. However, over the years, the value of predefining the target characteristics of the drug product is often underestimated. Consequently, the lack of a well-defined QTPP has resulted in wasted time and valuable resources. A recent paper by Raw et al.12 illustrates the significance of defining the correct QTPP before conducting any development. Also, QbD examples exemplify the identification and use of QTPPs.20–22
Product Design and Understanding
Over the years, QbD's focus has been on process design, understanding, and control, as discussed in the ICH Q8 (R2) guidance.3 It should be emphasized that product design, understanding, and control are equally important. Product design determines whether the product meets patients’ needs, which is confirmed by clinical studies. Product design also determines whether the product can maintain its performance through its shelf life, which is confirmed by stability studies. This type of product understanding could have prevented some historical stability failures.
The key objective of product design and understanding is to develop a robust product that can deliver the desired QTPP over the product shelf life. Product design is open-ended and may allow for many design pathways. Key elements of product design and understanding include the following:
- Physical, chemical, and biological characterization of the drug substance(s)
- Identification and selection of excipient type and grade, and knowledge of intrinsic excipient variability
- Interactions of drugs and excipients
- Optimization of formulation and identification of CMAs of both excipients and drug substance
To design and develop a robust drug product that has the intended CQAs, a product development scientist must give serious consideration to the physical, chemical, and biological properties of the drug substance. Physical properties include physical description (particle size distribution and particle morphology), polymorphism and form transformation, aqueous solubility as a function of pH, intrinsic dissolution rate, hygroscopicity, and melting point(s). Biopharmaceutical solid polymorphism, for example, has received much attention recently since it can impact solubility, dissolution, stability, and manufacturability. Chemical properties include pKa, chemical stability in solid state and solution, as well as photolytic and oxidative stability. Biological properties include partition coefficient, membrane permeability, and bioavailability.
Biopharmaceutical excipients are components of a drug product other than the drug substance. Excipients can:
- Aid in the processing of the dosage form during its manufacture
- Protect, support, or enhance stability, bioavailability, or patient acceptability
- Assist in product identification
- Enhance any other attribute of the overall safety, effectiveness, or delivery of the drug during storage or use.23
They are classified by the functions they perform in a biopharmaceutical dosage form. Among forty-two functional excipient categories listed in USP/NF,24 commonly used excipients include binders, disintegrants, fillers (diluents), lubricants, glidants (flow enhancers), compression aids, colors, sweeteners, preservatives, suspending/dispersing agents, pH modifiers/buffers, tonicity agents, film formers/coatings, flavors, and printing inks. The FDA’s inactive ingredients database25 lists the safety limits of excipients based on prior use in FDA-approved drug products.
It is well-recognized that excipients can be a major source of variability. Even though excipients can alter the stability, manufacturability, and bioavailability of drug products, the general principles of excipient selection are not well-defined, and excipients are often selected ad hoc without systematic drug-excipient compatibility testing. To avoid costly material wastage and time delays, ICH Q8 (R2) recommends drug-excipient compatibility studies to facilitate the early prediction of compatibility.3 Systematic drug-excipient compatibility studies offer several advantages as follows: minimizing unexpected stability failures which usually lead to increased development time and cost, maximizing the stability of a formulation and hence the shelf life of the drug product, and enhancing the understanding of drug-excipient interactions that can help with root cause analysis should stability problems occur.
Formulation optimization studies are essential in developing a robust formulation that is not on the edge of failure. Without optimization studies, a formulation is more likely to be high risk because it is unknown whether any changes in the formulation itself or the raw material properties would significantly impact the quality and performance of the drug product, as shown in recent examples.26,27 Formulation optimization studies provide important information on the following:
- Robustness of the formulation including establishing functional relationships between CQAs and CMAs
- Identification of CMAs of drug substances, excipients, and in-process materials
- Development of control strategies for drug substances and excipients
In a QbD approach, it is not the number of optimization studies conducted but rather the relevance of the studies and the utility of the knowledge gained for designing a quality drug product that is paramount. As such, the QbD does not equal the design of experiments (DoE), but the latter could be an important component of QbD.
Drug substances, excipients, and in-process materials may have many CMAs. A CMA is a physical, chemical, biological, or microbiological property or characteristic of an input material that should be within an appropriate limit, range, or distribution to ensure the desired quality of that drug substance, excipient, or in-process material.
Since there are many attributes of the drug substance and excipients that could potentially impact the CQAs of the intermediates and finished drug product, it is unrealistic that a formulation scientist investigate all the identified material attributes during the formulation optimization studies. Therefore, a risk assessment would be valuable in prioritizing which material attributes warrant further study. The assessment should leverage common scientific knowledge and the formulator’s expertise. A material attribute is critical when a realistic change in that material attribute can have a significant impact on the quality of the output material. Product understanding includes the ability to link input CMAs to output CQAs. The steps taken to gain product understanding may include the following:
- Identify all possible known input material attributes that could impact the performance of the product.
- Use risk assessment and scientific knowledge to identify potentially high-risk attributes.
- Establish levels or ranges of these potentially high-risk material attributes.s
- Design and conduct experiments, using DoE when appropriate
- Analyze the experimental data and, when possible, apply first principle models to determine if an attribute is critical
- Develop a control strategy. For critical material attributes, define acceptable ranges. For noncritical material attributes, the acceptable range is the range investigated. When more than one excipient is involved, these defined acceptable ranges may be termed formulation design space.
Process Design and Understanding
A biopharmaceutical manufacturing process usually consists of a series of unit operations to produce the desired quality product. Unit operations may be executed in batch mode in a continuous manufacturing process. A unit operation is a discrete activity that involves physical or chemical changes. A process is generally considered well-understood when;
- All critical sources of variability are identified and explained
- Variability is managed by the process
- Product quality attributes can be accurately and reliably predicted.28
Process parameters are referred to as the input operating parameters (e.g., speed and flow rate) or process state variables (e.g., temperature and pressure) of a process step or unit operation. A process parameter is critical when its variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired quality. Under this definition, the state of a process depends on its CPPs and CMAs of the input materials.
Specification
A specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a drug substance, drug product, or materials at other stages of its manufacture should conform to be considered acceptable for its intended use. “Conformance to specification” means that the drug substance and drug product, when tested according to the listed analytical procedures, will meet the acceptance criteria. Specifications are critical quality standards that are proposed and justified by the manufacturer and approved by regulatory authorities as conditions of approval. Specifications are one part of a total control strategy designed to ensure product quality and consistency. Other parts of this strategy include thorough product characterization during development, upon which many of the specifications are based, adherence to Good Manufacturing Practices, a validated manufacturing process, raw materials testing, in-process testing, stability testing, etc. Specifications are chosen to confirm the quality of the drug substance and drug product rather than to establish full characterization and should focus on those molecular and biological characteristics found to be useful in ensuring the safety and efficacy of the product.32
Justification of the specification:
- Specifications are linked to a manufacturing process
- Specifications should account for the stability of drug substance and drug product
- Specifications are linked to preclinical studies
- Specifications are linked to analytical procedures
Biopharmaceutical Development, Product Lifecycle, and Statistical Tools
Statistical methods demonstrate how biostatistics is used in many aspects of biopharmaceutical product development. The FDA perspective involving measurement system analysis, control charting, capability, acceptance sampling, and stability analysis along with the ICH guidelines are presented under the regulatory considerations for statistical analysis. Emphasis is given to areas where a biostatistician’s contribution will be crucial for following the critical path initiative of the FDA and collective effort by the biopharmaceutical industry in maintaining the highest level of quality and innovation, Figure 7.
To calculate the process capability index (Cpk), follow these steps:
- Define the specification limits for the process.
- Calculate the process mean (μ) and standard deviation (σˆ ).
- Determine the Upper and Lower Specification Limits (USL, LSL).
- Compute the Capability Index (Cpk) using the formula: Cpk = min [ (USL - μ) / (3σˆ ), (μ - LSL) / (3σˆ )].
Concept of Process Capability
- First introduced in the Statistical Quality Control Handbook by the Western Electric Company (1956).
- “Process capability” is defined as “the natural or undisturbed performance after extraneous influences are eliminated. This is determined by plotting data on a control chart.”
- ISO, AIAG, ASQ, and ASTM30 published their guideline or manual on process capability index calculation
Difference between Cpk and Ppk
- Cpk represents the potential process capability (i.e. how well a given process could perform when all special causes have been eliminated).
- Ppk addresses how the process has performed without the demonstration of the process to be stable.
- Forecast future batch failure rate
Where:
SD: overall variability, a standard deviation of all individual (observed) values, which accounts for both common cause variability (noise) and special cause variability. It is often referred to as overall variability.
σˆ (sigma hat): the inherent variability (noise) due to the common cause of a stable process. It is often estimated by using within-subgroup variability which is linked to the use of control charts.
USL: upper specification limit
LSL: lower specification limit
Mean: grand average of all the data
N: The number of data point
Xi: each of the values of the data
x- (X-Bar): the mean of xi
R- (R-Bar): The R- symbol is the arithmetic mean of the R values
Note: The inherent process variation should be estimated from control charts using a simple formula for, σˆ = R- /2, where R- is the average of the sample ranges, and d2 is a constant varying by sample size, which values can be found in the statistical table (d2 = 2.33 for a sample size of 5).
ASTM E2281: Standard Practice for Process and Measurement Capability Indices.
Cp: process capability index
Cpk: minimum process capability index
Pp: process performance index
Ppk: minimum process performance index
Calculation Formula
Cp =(USL-LSL)/6σˆ
Pp =(USL-LSL)/6SD
Cpkl=(Mean-LSL)/3σˆ
Ppkl=(Mean-LSL)/3SD
Cpku=(USL-Mean)/3σˆ
Ppku=(USL-Mean)/3SD
CpK=min (Cpkl, Cpku) or Cpk = min [ (USL−mean)/3σˆ , (mean−LSL)/3σˆ ]
Ppk= min (Ppkl, Ppku
A Perfectly Centered Process Mean
For this case:
USL= +4σ
LSL = -4σ
USL-LSL= 8σ
Cp = 1.333
Cpku=1.333
Cpkl=1.333
Cpk=1.333
For this case;
USL= +4σ
LSL = -4σ
USL-LSL= 8σ
Cp = 1.333
Cpkl= 1.667
Cpku= 1.0
Cpk= 1.0
- When the process is not centered, or deliberately run off-center for economic reasons, or only a single specification limit is involved, Cpk should be used.
- Similarly, Ppk offsets Pp weakness by introducing process mean in the calculation formula.
References
- Juran JM. Juran on quality by design: the new steps for planning quality into goods and services. New York: The Free Press; 1992. [Google Scholar]
- Woodcock J. The concept of Biopharmaceutical quality. Am Pharm Rev 2004; 1–3.
- U. S. Food and Drug Administration. Guidance for Industry: Q8 (2) Biopharmaceutical Development. 2009
- U. S. Food and Drug Administration. Guidance for Industry: Q9 Quality Risk Management. 2006.
- U. S. Food and Drug Administration. Guidance for Industry: Q10 Biopharmaceutical quality system. 2009.
- U. S. Food and Drug Administration. Guidance for Industry: Q8, Q9, and Q10 questions and answers. 2011.
- ICH Quality Implementation Working Group. Points to consider. ICH-endorsed guide for ICH Q8/Q9/Q10 implementation. 2011.
- U. S. Food and Drug Administration. Guidance for Industry: Q11 development and manufacture of drug substance. 2012.
- U. S. Food and Drug Administration. FDA-EMA parallel assessment of Quality-By-Design elements of marketing applications. http://www.fda.gov/Drugs/DrugSafety/ucm365524. htm. Accessed 16 Nov 2013
- Yu LX. Biopharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25:781–91. doi: 10.1007/s11095-007-9511- 1. [PubMed] [CrossRef] [Google Scholar]
- Lionberger R, Lee S, Lee L, Raw A, Yu LX. Quality by design: concepts for ANDAs. AAPS J. 2008;10:268–76. Doi: 10.1208/s12248-008-9026-7. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- Raw AS, Lionberger R, Yu LX. Biopharmaceutical equivalence by design for generic drugs: modified-release products. Pharm Res. 2011;28:1445–53. doi: 10.1007/s11095-011- 0397-6. [PubMed] [CrossRef] [Google Scholar]
- Rathore AS, Winkle H. Quality by design for biopharmaceuticals. Nat Biotechnol. 2009;27:26–34. doi: 10.1038/nbt0109-26. [PubMed] [CrossRef] [Google Scholar]
- U. S. Food and Drug Administration. Guidance for Industry: tablet scoring: nomenclature, labeling, and data for evaluation. 2013.
- U. S. Food and Drug Administration. Guidance for Industry: Size of Beads in Drug Products Labeled for Sprinkle. January, 2011.
- U. S. Food and Drug Administration. Summary Minutes of the Advisory Committee for Biopharmaceutical Science and Clinical Pharmacology. July 26, 2011. http://www. fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/ AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM272111.pdf. Accessed 13 Aug 2013.
- U. S. Food and Drug Administration. Guidance for industry: immediate release solid oral dosage forms scale-up and Post-approval changes: chemistry, manufacturing, and controls, in vitro dissolution testing, and in vivo bioequivalence documentation. 1995.
- U. S. Food and Drug Administration. Guidance for industry: modified release solid oral dosage forms scale-up and Post-approval changes: chemistry, manufacturing, and controls, in vitro dissolution testing, and in vivo bioequivalence documentation. 1997.
- U. S. Food and Drug Administration. Guidance for industry: CMC post-approval manufacturing changes are to be documented in annual reports. 2014.
- U. S. Food and Drug Administration. Quality by design for ANDs: an example for immediate-release dosage forms. 2012. http://www.fda.gov/downloads/ Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM304305.pdf. Accessed 16 Nov 2013.
- U. S. Food and Drug Administration. Quality by design for ANDs: an example for modified-release dosage forms. 2011. http://www.fda.gov/downloads/ Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM304305.pdf. Accessed 16 Nov 2013.
- CMC Biotech Working Group. A Mab: A case study in bioprocess development. http://www. google.com/url?sa=t&rct=j&q=&esrc=s&frm=1&source=web&cd=8&ved=0CF8QFjA H&url=http%3A%2F%2Fwww.ispe.org%2Fpqli%2Fa-mab-case-study-version2.1&ei=f 6qIUpXqKuLlsATlioHgDA&usg=AFQjCNH9JB17H4gssCd489dlOwV4xoAmDQ. Accessed 16 Nov 2013.
- USP 34—NF 29 (United States Pharmacopeial Convention). Chapter 1078. Good manufacturing practice for bulk Biopharmaceutical excipients. Rockville, MD: USP; 2011, pp. 1415–1420
- USP 34—NF 29 (United States Pharmacopeial Convention). USP and NF Excipients, Listed by Category. Rockville, MD: USP; 2011, pp. 583–595.
- U. S. Food and Drug Administration. Inactive Ingredient Search for Approved Drug Products. http://www.accessdata.fda.gov/scripts/cder/iig/index.cfm, Accessed 13 Aug 2013.
- Nazzal S, Nutan M, Palamakula A, Shah R, Zaghloul AA, Khan MA. Optimization of a self-nanoemulsified tablet dosage form of ubiquinone using response surface methodology: effect of formulation ingredients. Int J Pharm. 2002;240:103–14. doi: 10.1016/S0378- 5173(02)00130-8. [PubMed] [CrossRef] [Google Scholar]
- Awotwe-Otoo D, Agarabi C, Wu GK, Casey E, Read E, Lute S, et al. Quality by design: impact of formulation variables and their interactions on quality attributes of a lyophilized monoclonal antibody. Int J Pharm. 2012;438(1–2):167–75. doi: 10.1016/j. ijpharm.2012.08.033. [PubMed] [CrossRef] [Google Scholar]
- U.S. Food and Drug Administration CDER. Guidance for industry: PAT—a framework for innovative Biopharmaceutical development, manufacturing, and quality assurance. 2004.
- NIST/SEMATECH e-Handbook of Statistical Methods. What is process capability? http:// www.itl.nist.gov/div898/handbook/pmc/section1/pmc16.htm. Accessed on 13 Aug 2013.
- ASTM E2281—08a (2012)e1 Standard practice for process and measurement capability indices. http://www.astm.org/Standards/E2281.htm. Accessed 13 Aug 2013.
- ICH Q6A specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances - Scientific guideline, May 2000, CPMP/ ICH/367/96; https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-6-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products-chemical-substances-step-5_en.pdf
- Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products, U.S. Department of Health and Human Services Food and Drug Administration, August 1999, ICH; Q6Bfnl.PDF (fda.gov).
- Lawrence X. Yu, Gregory Amidon, Mansoor A. Khan, Stephen W. Hong, James P. Polli, G. K. Raju, and Janet Woodcock; Understanding Pharmaceutical Quality by Design, AAPS J. 2014 Jul; 16(4): 771–783. Published online 2014 May 23. doi: 10.1208/s12248-014-9598-3, PMCID: PMC4070262PMID: 24854893; Understanding Pharmaceutical Quality by Design - PMC (nih.gov)
Author Details
Robert Dream - HDR COMPANY LLC
Publication Details
This article appeared in American Pharmaceutical Review: Vol. 27, No. 6Sept/Oct 2024Pages: 40-50
Subscribe to our e-newsletters.
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!