Introduction
The authors have become aware that regulators, especially during site inspections, are beginning to ask questions about whether pharmaceutical manufacturers have considered setting time limits from the date of manufacturing to the microbial release testing of their finished drug products. Although good manufacturing practice regulations, guidance documents, testing manuals, and pharmacopeial chapters discuss sampling and method validation issues, generally they are silent on the timing of the microbial testing of sterile and non-sterile drug products. Is this absence of guidance an oversight or is this not considered an important issue, with release testing driven by scheduling and a need to release the drug product to the market as soon as is practically possible? We believe it is the latter. As the survival or proliferation of microorganisms is dynamic, unlike most other test parameters, e.g., potency, the question is worth asking and may be a consideration for all microbial testing throughout the product life cycle as well as at product release. Sterility testing is not conducted on drug products subject to parametric release and some cellular blood[1]derived biologicals, which are exempt from the 21 CFR 610.12 Sterility requirements.
This brief review will summarize the regulations, discuss the possible implications of the timing of microbial testing on product quality and patient safety and provide case studies related to the issue.
Good Manufacturing Practice Regulations
The U.S. Federal Good Manufacturing Practice regulations 21 CFR 211. 165 Testing and release for distribution states:
a. For each batch of drug product, there shall be appropriate laboratory determination of satisfactory conformance to final specifications for the drug product, including the identity and strength of each active ingredient, prior to release. Where sterility and/or pyrogen testing are conducted on specific batches of short-lived radiopharmaceuticals, such batches may be released prior to completion of sterility and/or pyrogen testing, provided such testing is completed as soon as possible.
For biological products, 21 CFR 611.12 Sterility is the applicable regulation, which states:
a. The test. Except as provided in paragraph (h) of this section, manufacturers of biological products must perform sterility testing of each lot of each biological product’s final container material or other material, as appropriate and as approved in the biologics license application or supplement for that product.
b. Test requirements.
- The sterility test must be appropriate to the material being tested such that the material does not interfere with or otherwise hinder the test.
- The sterility test must be validated to demonstrate that the test is capable of reliably and consistently detecting the presence of viable contaminating microorganisms.
- The sterility test and test components must be verified to demonstrate that the test method can consistently detect the presence of viable contaminating microorganisms.
c. Written procedures. Manufacturers must establish, implement, and follow written procedures for sterility testing that describe, at a minimum, the following:
1. The sterility test method to be used.
(i) If culture-based test methods are used, include, at a minimum:
- Composition of the culture media.
- Growth-promotion test requirements; and
- Incubation conditions (time and temperature).
(ii) If non-culture-based test methods are used, include, at a minimum:
- Composition of test components.
- Test parameters, including acceptance criteria; and
- Controls used to verify the method’s ability to detect the presence of viable contaminating microorganisms.
2. The method of sampling, including the number, volume, and size of articles to be tested.
3. Written specifications for the acceptance or rejection of each lot; and
4. A statement of any other function critical to the sterility test method to ensure consistent and accurate results.
d. The sample. The sample must be appropriate to the material being tested, considering, at a minimum:
- The size and volume of the final product lot.
- The duration of manufacturing of the drug product.
- The final container configuration and size.
- The quantity or concentration of inhibitors, neutralizers, and preservatives, if present, in the tested material.
- For a culture-based test method, the volume of test material that results in a dilution of the product that is not bacteriostatic or fungistatic; and
- For a non-culture-based test method, the volume of test material that results in a dilution of the product does not inhibit or otherwise hinder the detection of viable contaminating microorganisms.
The 2004 FDA Guidance for Industry Sterile Drugs Produced by Aseptic Processing – Current Good Manufacturing Practice states:
Certain aspects of sterility testing are of particular importance, including control of the testing environment, understanding the test limitation, and investigating manufacturing systems following a positive test.
The EU sterility test requirements are found in the Good Manufacturing Practices – Annex 1: Manufacture of Sterile Products, which states:
10.5 The sterility test applied to the finished product should only be regarded as the last in a series of control measures by which sterility is assured. It cannot be used to assure sterility of a product that does not meet its design, procedural, or qualification parameters. The test should be validated for the product concerned.
10.6 The sterility test should be performed under aseptic conditions. Samples taken for sterility testing should be representative of the whole of the batch but should in particular include samples taken from parts of the batch considered to be most at risk of contamination, for example:
- For products that have been filled aseptically, samples should include containers filled at the beginning, middle,e and end of the batch and after any significant intervention (e.g., interventions where the integrity of a barrier is breached (open door)) or an operator intervention into critical zones.
- For products that have been heat sterilized in their final containers, samples taken should be representative of the worst-case locations (e.g., the potentially coolest or slowest to heat part of each load).
- For products that are lyophilized, samples are taken from different lyophilization loads.
10.7 For some products it may not be possible to perform a sterility test prior to release because the shelf life of the product is too short to allow completion of a sterility test. In these cases, the CCS should clearly capture the identified risks, the additional considerations of design of the process, and additional monitoring required to mitigate the identified risks.
The authors believe it is telling that none of the four salient regulatory documents cited above address the timing of the finished product testing. If this were an important issue, we would have expected these regulations to contain instructions for this requirement.
Other Guidelines Addressing Sterility Testing
FDA Pharmaceutical Microbiology Manual
This manual, which was written by expert FDA microbiologists (that included one of our authors) merely states:
Samples are to be held under the same storage conditions as required by the package label or insert.
FDA Compliance Program 7356.002M for Inspection of Large Molecule Products
The role of this document is the training of FDA field investigators, and it states:
5. Laboratory Control System
A. All Laboratory Disciplines Supplemental question: Are the firm’s laboratory analysts and management staff qualified to analyze, review, and evaluate data and quality assurance/quality control requirements?
(1) Sampling: Does the firm have procedures for raw material sampling and testing? Is the firm’s raw material sampling adequate to ensure proper conclusions on raw material disposition?
Supplemental questions:
- Is the firm’s in-process material sampling consistent with the sampling described in relevant BLAs?
- Is the firm’s in-process sampling appropriate for monitoring the manufacturing process?
- Are the firm’s sampling plans for protein DS release testing consistent with sampling plans defined in relevant BLAs and representative of the batch being tested?
- Does the firm have adequate sample tracking?
- Is an adequate system in place to ensure that samples are stored appropriately and that the correct samples are tested within appropriate time frames specified in SOPs?
- Does the firm store test samples under conditions that prevent stress or destruction?
- Are microbial bioburden and endotoxin samples stored at 2–8° C for less than 24 hours?
- If other storage conditions are used for bioburden and endotoxin samples, are data available to demonstrate that recovery/viability is not compromised by the storage conditions?
(2) Test Methods: Are the firm’s test methods consistent with those described in relevant BLA
- Do the acceptance criteria match those described in relevant applications? Supplemental question: Does the firm have adequate SOPs for each assay?
Do assays have adequate system suitability criteria?
The statement above in bold font that states “the correct samples are tested within appropriate time frames specified in SOPs” is ambiguous. This agency guidance does not define “correct samples” in this sentence. Since it is open to interpretation, those of us who are Industrial Microbiologists would view the term “correct samples” to mean those that are associated with pre-filtration bioburden, water samples, and sterilization Biological Indicators. We believe this listed question directs the investigator to review the procedural instructions for the more susceptible samples that are vulnerable to improper time and temperature storage conditions. Those samples associated with sterility testing product release are understood to have patient urgency and common-sense practices have those products released as soon as they are acceptable. Does it mean that manufacturers must have microbial testing time frames, or do they comply with their SOPs? However, this compliance training document with inspectional reporting requirements may be the origin of the investigator’s questions without providing a definitive answer.
Pharmacopeial Chapters
Pharmacopeial standards are meant to enhance consistency in testing and provide the appropriate quality criteria. The most referenced USP chapters for microbiological quality analysis are <61> Microbiological Examination of Non-sterile Drug Products: Microbial Enumeration Tests, <62>, Microbiological Examination of Non-sterile Drug Products: Tests for Specified Microorganisms and <71> Sterility Testing. Each of these chapters states the pharmacopeial perspective for the number or volume of samples tested but does not discuss methods or parameters of sampling, other than the use of an aseptic technique. USP <1117> Microbiological Good Laboratory Practices became an official general information chapter to offer some guidance supporting the procedures for microbiological quality analysis since numerous parameters involved in microbiological analysis are at best based on historical experiences that attempt to reduce inherent variability that is part of the science of microbiology. USP <1117> states:
“Good practices in a microbiology laboratory consist of activities that depend on several principles: aseptic technique, control of media, control of test strains, operation and control of equipment, diligent recording and evaluation of data, and training of the laboratory staff in related competencies. Because of the inherent risk of variability in microbiology data, reliability and reproducibility are dependent on the use of accepted methods and adherence to good practices in the laboratory.”
The only indication of sample handling and testing consistency is referred to as “training of laboratory staff in related competencies.
Specifically, though, in <1117> the section Sample Handling indicates that since samples are stored often after sampling,
… it is important to minimize the amount of time between the sampling event and the initiation of testing….
The latter is related to storage or transport conditions and further text encourages control of those conditions to ensure there is no impact on recovering microorganisms in the sample. One example of time between sampling and testing is given for bioburden testing of water, which originated from the US Environmental Protection Agency guidelines for water testing (since water samples are often taken remotely and transported to an EPA laboratory for testing). The water bioburden timeline is 24 hours between sampling, holding at 2-8°C, and testing. Where greater than 24 hours occur, it is recommended that supporting experimental studies show a maximum hold time allowance. It is important to recognize that water bioburden testing parameters from the EPA relate to potable water with varying levels of bioburden, thus non-sterile material.
Also, environmental monitoring microbiology samples “if…cannot be incubated within a reasonable time frame…the time from sample collection to the start of incubation should be supported with experimental data.”
These pharmacopeial recommendations indicate that a standard specific time allowance between sample collection and sample testing is not clear-cut. Knowledge of the aseptic or terminally sterilized operation, the formulation of the sample (e.g., does it contain antimicrobial properties or ingredients), and any hold or storage conditions relevant prior to testing, are all significant contributors to the potential recovery probability and should be part of a risk assessment related to time allowance for testing on a product basis. For example, a terminally sterilized product should not need a time allowance for sterility testing. Prefiltration bioburden samples could have detectable microbial recoveries and may need time guardrails added to the firm’s laboratory SOP to ensure the accuracy of that testing. Concomitant with determining if time allowance is necessary is the reality that sterility testing is not a guarantee that 100% of units will be sterile. The latter is fraught with variability, lack of sensitivity, and low contamination detectability based on probability. The pharmacopeial (USP) guidance is reasonable and leaves the determination of time allowance to the competency of laboratory personnel responsible for sterility testing and who best know their products.
General Discussion of the Time Frame for Microbial Testing
Sterile Drug Products
Exceptions to the requirement to complete the sterility test prior to product release are the short-lived products which include radiopharmaceuticals, and cell and gene therapies where batches are released at risk and may be administered to the patient prior to the completion of the test, terminally sterilized sterile injectable products subjected to parametric release, and a limited range of blood-derived products, which are exempt from sterility testing (21 CFR 610.12 Sterility). There are three scenarios with a potentially contaminated sterile drug product. If the sample for sterility testing contains no viable microorganisms, in the absence of a laboratory error, the batch will meet the requirements of the test and may be released for distribution. Alternatively, if the sample contains viable microorganisms, during the time from manufacture to testing, the microbial contaminant may die off, persist, or proliferate within the sample. If the microorganism dies off, then the batch will pass the test and will be suitable for administration; if the microorganism persists or proliferates, then the batch will fail the test and will be rejected. These outcomes are largely independent of the hold time as the limit of detection of a compendial sterility test is expected to be as low as 1 colony-forming unit.
Other requirements that trigger sterility testing are the annual or end-of-shelf-life sterility testing (versus container-closure integrity testing) in a long-term stability program or the testing of retained samples from a batch in the field by the manufacturer, and/ or regulatory agency acting in response to a clinical complaint that a product is contaminated, which would all lie outside any proposed hold time for release testing. Would this mean that an FDA regional microbiology laboratory would be using an invalid sterility test when testing suspect product containers collected from pharmacy stock shelves or hospitals when investigating an infection outbreak if it were tested outside a proposed company hold time? This is not the case because the FDA and other health authorities for decades have tested both for-cause and surveillance product samples to monitor the sterility of commercially available products throughout their shelf life.
Sterile lyophilized drug products are a special case. The presence of cryoprotectants and bulking agents within the formulation and storage at refrigeration temperature to protect the stability of the active ingredient during the shelf life will also protect a microbial contaminant so that the timing of the sterility test will be largely irrelevant.
Non-Sterile Drug Products
Non-sterile drug products have microbial enumeration limits and the absence of specified microorganism requirements based on the specific microbial quality attributes delineated in the USP monograph for these types of products and reflect the risk to the recipient based on the product attributes and the invasiveness of the route of administration. Product characteristics like extreme pH, low water activity, and antimicrobial activity are inimical to the survival of microorganisms so microbial contaminants may die off with time. For example, antimicrobial preservative systems must meet the USP <51> Antimicrobial Effectiveness Test log reduction requirements for their dosage form requirements at 7, 14, and 28 days after inoculation with a challenge microorganism. Although the addition of an antimicrobial agent is intended to protect a multi-use drug product during its shelf life and not to eliminate contaminating microorganisms due to poor GMP practices microorganisms within the product will be reduced by a delay in testing from manufacturing to testing. Due to this scenario, the microbial counts may decline from immediately after manufacture to the scheduling of the microbial testing. This will have no consequence to patient safety as the testing immediately prior to product release will be most representative of the bioburden in the product during the shelf life.
In-Process Bioburden Samples
Samples taken during downstream processing of bio-pharmaceuticals or pre-sterile filtration bulk drug products have more relevant storage and hold time requirements than finished drug products. Every attempt should be made to ensure the microbial plate count results are indicative of the bioburden prior to the next processing step. Sample storage at refrigeration temperature and testing as soon as practically possible will prevent the bioburdenfrom changing during the time period between sampling and microbial enumeration.
Case Study
The below case study illustrates the difficulties in conducting studies necessary for applying a sample hold-time before sterility testing. It details an experiment where less than 100 CFU of various microorganisms were added to three batches of a biological drug and three Fluid A replicates – a common diluent in sterility test methods, which served as a control. To compare, samples were tested immediately or after being stored at 2-8°C for six and twelve days.
Upon completion of the sterility testing, the samples were placed into incubation for a period of up to 14 days, or until evidence of turbidity indicated microbial growth. Observations were noted as either growth or no growth. Data is presented in Tables 1-3. The results demonstrated microbial growth of all inoculated organisms except for some replicates of Roseomonas mucosa at time zero hold. This inability to consistently cultivate Roseomonas mucosa was observed for the biological drug product and Fluid A, suggesting that the lack of growth was not related to the specific makeup of the product. When testing occurred after a six-day hold time, there were failures to grow Candida albicans, Cladosporium species, and Roseomonas mucosa within the product, as well as Pseudomonas aeruginosa, Cladosporium species, and Roseomonas mucosa in Fluid A. When testing occurred after a twelve-day hold time, there were failures to grow Cladosporium species and Roseomonas mucosa in the biological drug product observed, and Pseudomonas aeruginosa, Staphylococcus aureus, and Roseomonas mucosa in Fluid A.
As shown in the case study, producing results is expensive and time-intensive without ensuring all microorganism recovery or yielding beneficial data for managing a pharmaceutical microbiology lab. The biological product in question was similar to control Fluid A regarding organism survival during hold time. Thus, conducting individual hold-time studies may not yield definitive outcomes. Also, some microorganisms didn’t grow even with no hold time, indicating that no specific duration guarantees their consistent recovery.
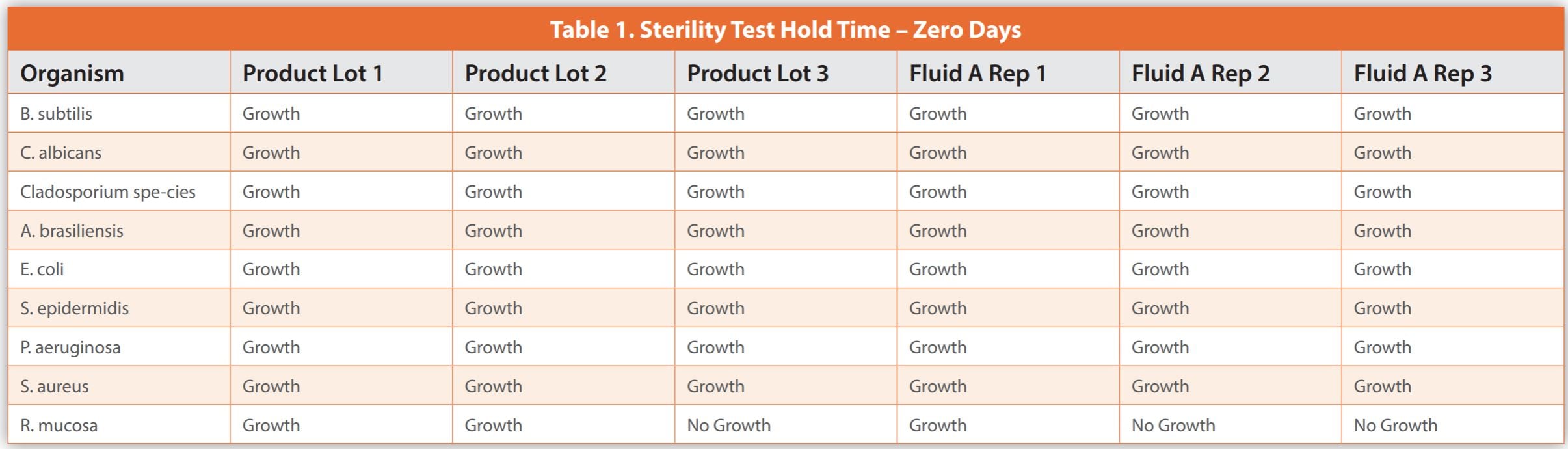
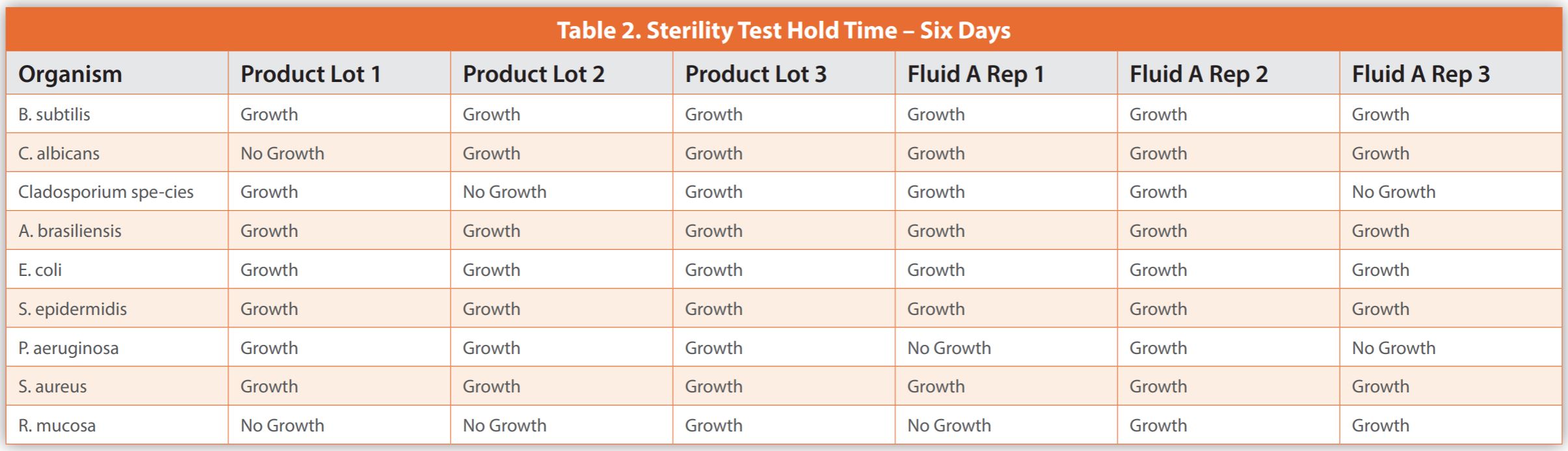
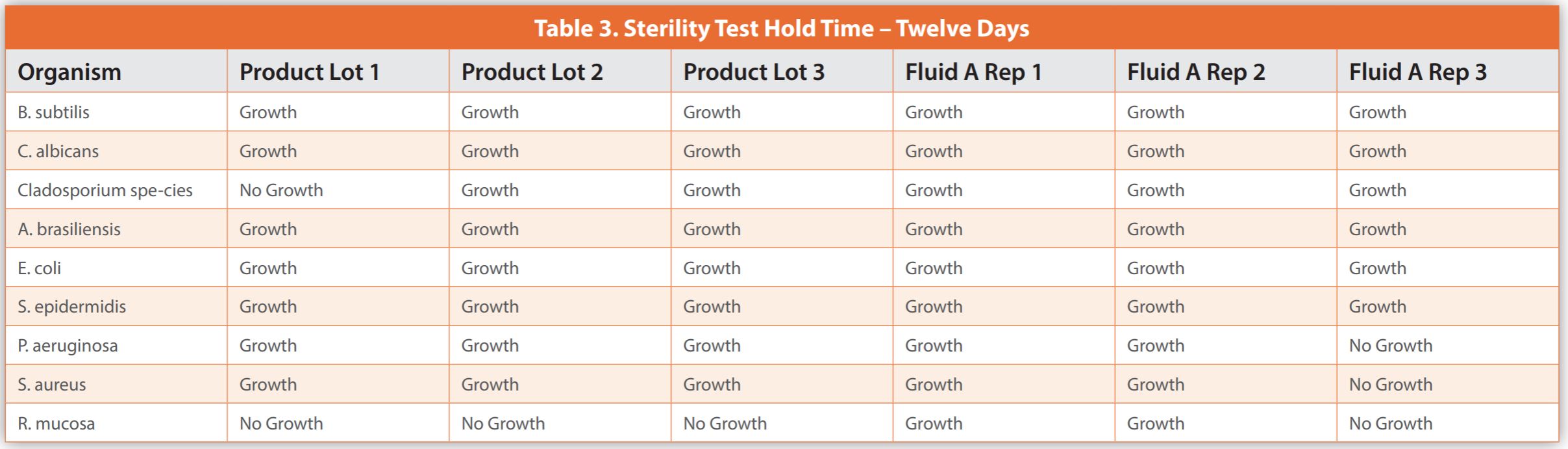
The data from the case study, while not large enough to be statistically broad-based, does not demonstrate that testing the biological product within six days is superior to 12 days for microorganism recovery. Testing after no sample hold time did recover more organisms, but still failed to recover Roseomonas mucosa, and would not be practical for most laboratories. The variability in results makes qualifying a product hold time very difficult and not scientifically sound. The most practical implementation of the discoveries from this case study would be to organize the laboratory so that samples for sterility are tested as soon as possible and in a first-in-first-out manner. The laboratory should have capacity and resources so that there is not an unreasonable backlog of sterility samples.
Conclusions
As opinion leaders in microbiology in the pharmaceutical industry, the authors strongly support the necessity to maintain Current Good Manufacturing Practices, especially with the advent of new products such as cell and gene therapies, greater barriers between human operators and aseptically processed drug products, and use of emerging microbiological testing technologies. As the GMP regulations first introduced in 1977 are legal requirements and are relatively static, we expect that how the GMPs can be implemented will be addressed in Guidance for Industry documents and not 483 observations or podium presentations. In contrast to FDA internal compliance documents, these guidelines are published, reviewed, and subject to comments from industry, and revised by the regulators. The introduction of new GMP requirements during FDA site inspections, without prior industry review, may not be based on sound scientific principles or established industry practices.
On reflection, the authors see no justification for regulators to push for the implementation of time frames for sterility testing for product release, rather, testing should be completed as soon as practical and with relevant priority.
References
- 21 CFR 211. 165 Testing and release for distribution
- 21 CFR 611.12 Sterility
- FDA Guidance for Industry Sterile Drugs Produced by Aseptic Processing – Current Good Manufacturing Practice 2004
- E.U. Good Manufacturing Practices – Annex 1: Manufacture of Sterile Product
- FDA Pharmaceutical Microbiology Manual 20 August 2020
- FDA Compliance Program 7356.002M for Inspection of Large Molecule Products
- USP <71> Sterility Tests
- USP <61> Microbiological Examination of Non-sterile Drug Products: Microbial Enumeration Tests
- USP <62> Microbiological Examination of Non-sterile Drug Products: Tests for Specified Microorganisms
- USP <1117> Microbiological Good Laboratory Practices
Author Details
Tony Cundell - Microbiological Consulting, LLC, Rye, New York; Phil Duncanson - AstraZeneca, Newark, Delaware; Dennis Guilfoyle - Johnson & Johnson, New Brunswick, New Jersey; Don Singer - Eco Lab, St. Paul, Minnesota.
Publication Details
This article appeared in American Pharmaceutical Review:Vol. 27, No. 7Nov/Dec 2024Pages: 8-13
Subscribe to our e-Newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!