Peter Tattersall, Lianjia Ma, Qinggang Wang, Li Li, and Jonathan Shackman- Chemical Process Development, Bristol Myers Squibb
Abstract
Active pharmaceutical ingredient process development requires comprehensive analytical controls utilizing a wide range of analytical methods. An analytical risk survey program was previously developed to evaluate common risk factors in the analytical control strategy for early to mid-stage development. For late-stage and the development of commercial methods, the critical quality-impacting analytical control requirements for manufacturing processes are typically captured in an analytical target profile. Given that every project is unique, how do we ensure consistency between projects in handling and learning from method risk evaluation and mitigation?
To address this, a novel analytical risk assessment program was implemented at Bristol Myers Squibb. In this article, we introduce a simple and widely applicable workflow to assess gaps and risks for specific methods being developed for commercial use that goes more in-depth than the survey program described previously. The workflow primarily utilizes spreadsheets, containing simple predefined lists of potential method concerns, to evaluate critical method parameters that may impact routine method performance and robustness in a QC setting. The templated format allows for more uniform reviews and lends to more efficient discussions around remaining method risk. We will explain the benefits of this risk assessment program over seven years for both the analytical development team and commercial stakeholders.
Introduction
A comprehensive set of analytical controls is required for active pharmaceutical ingredient (API) processes at the late-stage development phase, which typically utilizes a wide range of analytical techniques and methods. The analytical methods to support manufacturing processes at the commercial stage are developed and optimized based on process knowledge and analytical target profile (ATP) requirements. While some methods may be complementary, many cases require novel, specifically developed methods to ensure selectivity, sensitivity, precision, and accuracy for assay and impurity tests. These methods may be used for registrational starting materials, in-process controls (IPCs), intermediates, and the API itself.
In preparation for commercial process validation, also known as process performance qualification (PPQ), it is critical to have knowledge regarding specific method performance and robustness ahead of formal method validation. Establishing robust and reproducible test methods ahead of internal commercial manufacture or at contract manufacturing organizations (CMOs) is paramount to guarantee consistent performance and quality data, ensuring patient safety and enabling effective tracking and trending of data. To evaluate specific methods, we need a tool that can be widely applied to assess conditions and performance, identify knowledge gaps, and mitigate risks. Analytical risk assessments (RAs), based on the principles of ICH Q8, Q9, Q10, and Q11, 1- 4 should have the scope and depth to determine if methods are fit for purpose (FFP), i.e., simple, robust, and efficient. At Bristol Myers Squibb (BMS), an analytical RA program was developed to assess both common and less common risk factors for different types of methods. Figure 1 summarizes these assessment activities along the development timeline to commercialization, also showing that the amount of resources and eff ort around these activities necessarily increase. Note, to complement the in-depth, comprehensive RA program for late-stage API processes, an analytical risk survey workflow was previously developed to evaluate common risk factors in the analytical control strategy for early to mid-stage development.5
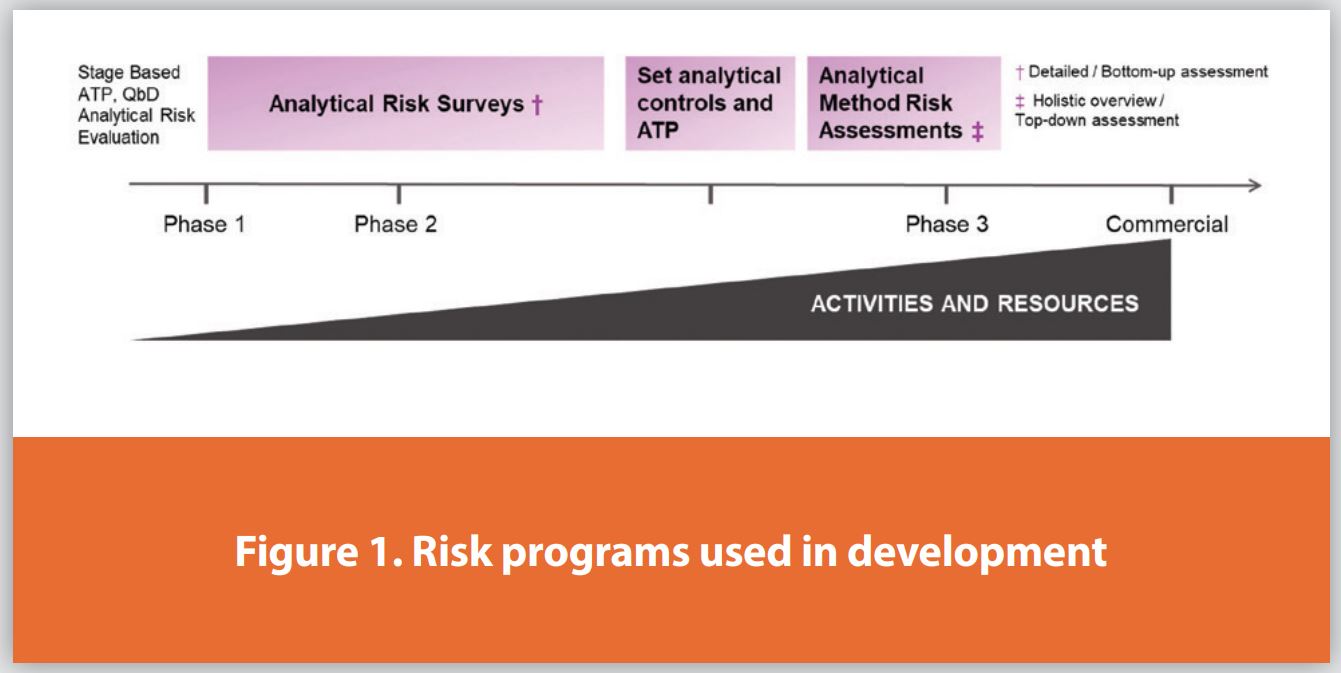
Our analytical RA program focuses on specific (non-general or non-compendial) analytical methods and their connection to process control needs, required acceptance criteria, and output data quality. This RA is designed to be performed on what are considered the final, optimized method parameters with established conditions (ECs) to confirm readiness for registrational validation studies. It uses a simple workflow that is widely applicable to assess analytical gaps and risks for methods routinely used in a QC lab setting. It incorporates a detailed, ‘bottom-up’ assessment for specific method and technique types. When performed with input from subject matter experts, the RA highlights areas of concern by combining a checklist of common risk factors with a spreadsheet for ranking the risk severity. Beyond just ranking risk, this facilitates the identification of knowledge gaps and the creation of an experimental ‘to-do’ tracker list for evaluating the analytical method conditions and optimizing controls. Once the method evaluation is complete and concerns are addressed (i.e., knowledge gathering experiments executed and identified risks are accepted or mitigated), the RA ultimately confirms readiness for commercial validation.
Discussion
ICH and method development workflow
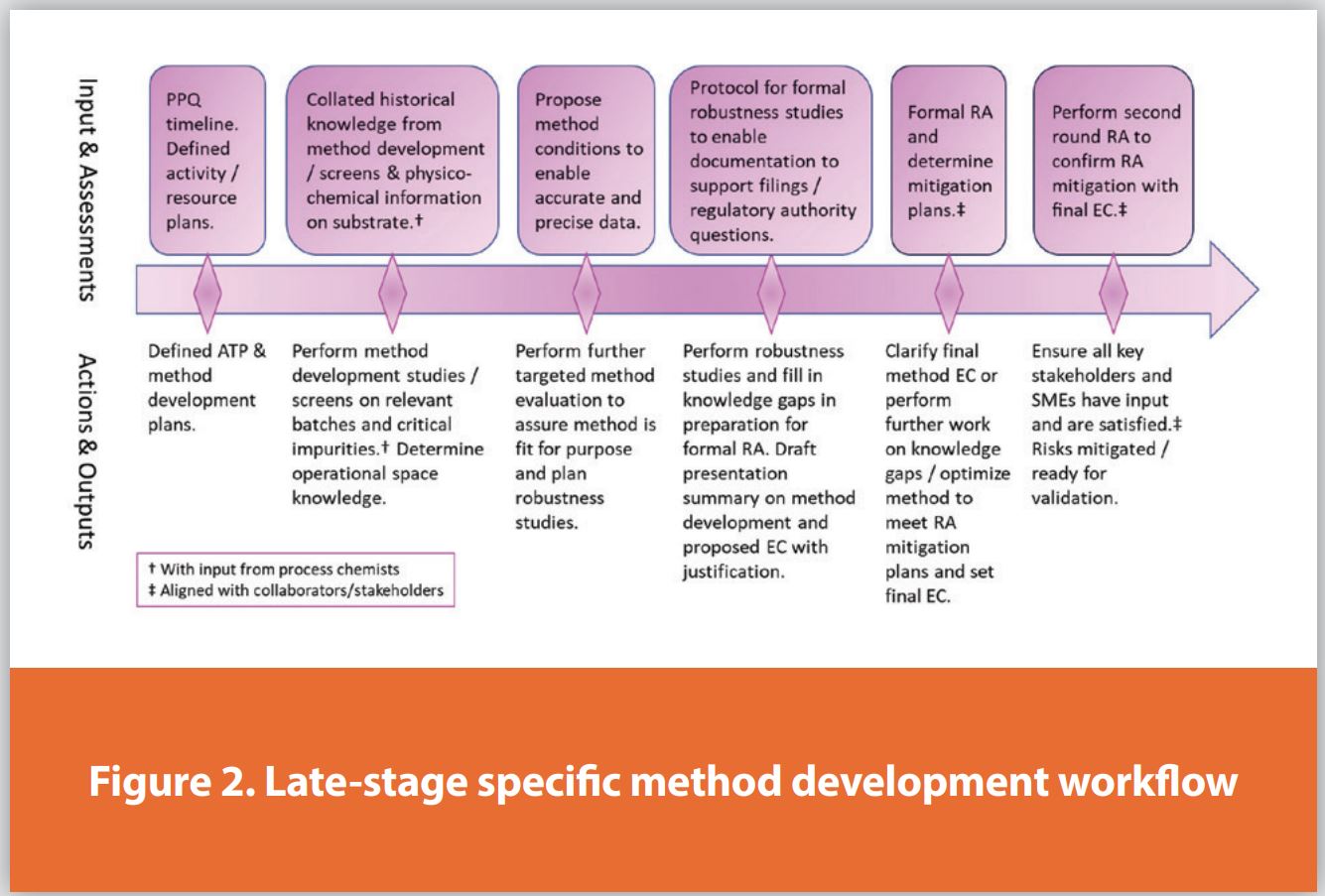
In recent years, there has been a focus on introducing quality by design (QbD) development approaches in line with ICH Q8, Q9, and Q10, mainly targeting API synthesis and final formulation. In 2022, ICH updated Q2(R2) and released Q14,6,7 providing guidance primarily aimed at registrational filings. Q14 aligns the analytical testing method needs with QbD principles. Commercial methods are developed and optimized based on the critical, quality-impacting analytical control needs for finalized commercial manufacturing processes as captured in the ATP. Method validation is necessary before the technical transfer to commercial QC labs in preparation for the PPQ campaign. To ensure our practices aligned with ICH Q2(R2) and Q14 guidance, we evaluated our late-stage method development approach. Our workflows were updated to incorporate final method optimization, formally documented robustness studies, and RA, all with QbD as the underlying driver. The event workflow for specific registrational methods was updated with the addition of targeted method evaluation control actions to guide development progress, as shown in Figure 2. At each checkpoint, the existing knowledge is assessed to determine the probability of success, and that the method is performing to phase-appropriate expectations.
Performing a Risk Assessment Method
RAs are a valuable component of the method development journey and decision-making process. They require a significant amount of up-front knowledge gathering, preparation, and presentation to meet the desired goals. Therefore, RAs are not recommended for all methods; only specific and challenging methods justify this level of effort. To obtain the optimum value and benefit from the RA, it is important to balance timing, presentation format, and contributors. RA participants generally include the method developer (author), analytical project lead, analytical RA subject matter experts (SMEs), meeting coordinator, and identified stakeholders such as quality and commercial analytical representatives. The associated synthetic chemist and/or chemical engineer may also be included optionally.
The author typically collates the method development history, ATP, and validation/specification requirements. Presentations are based on a templated approach covering analyte physicochemical information, method challenges with relevant data, historical/upcoming key decisions, operating space/parameter ranges, and robustness study data, all within a presentation slide deck to ‘set the scene.’ The method developer is also asked to collate method knowledge and information by populating a method risk assessment spreadsheet template based on the proposed method. Additionally, for context, the project tasks, timeline (based on process characterization, validation deadlines, technical transfer plans, customer timelines, and stability studies), and method criticality to the control strategy are outlined.
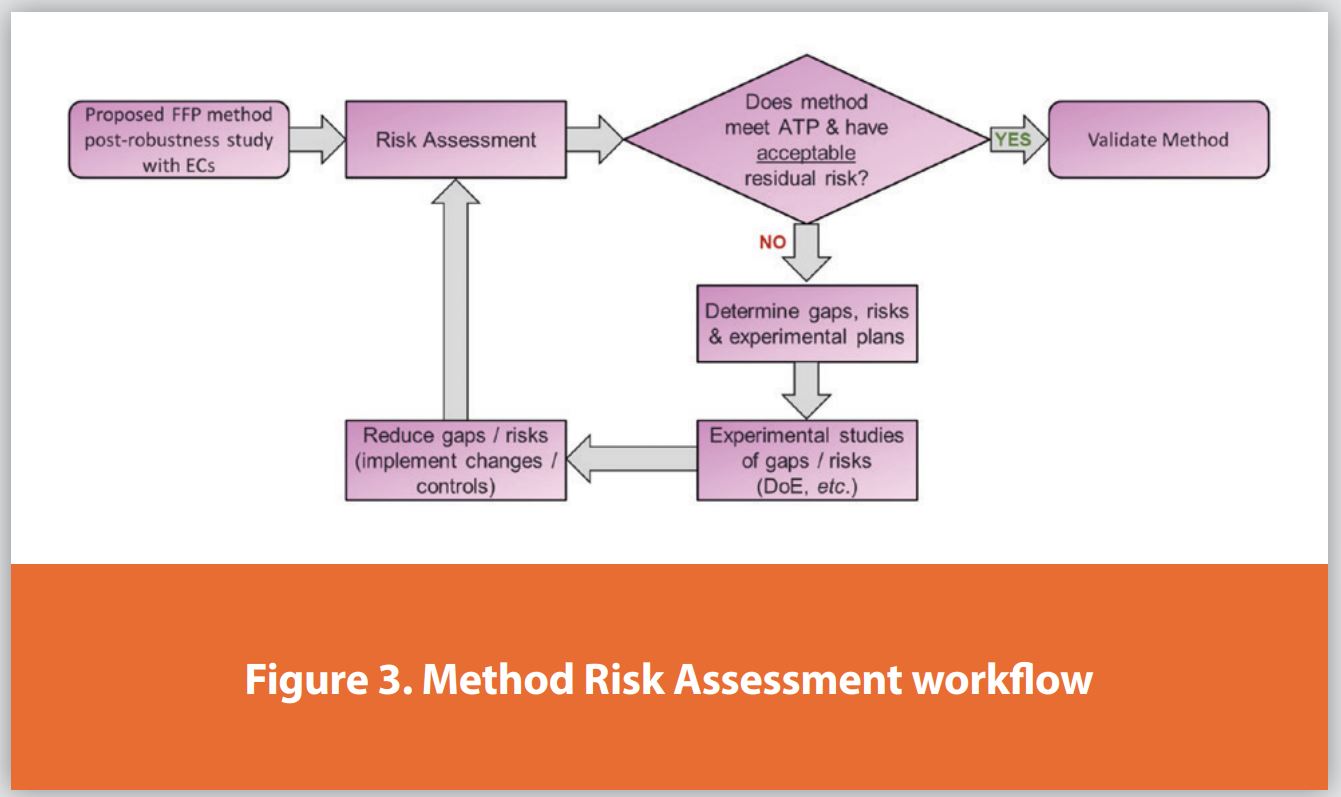
The RA workflow, where the appropriateness of the method,d technique, and ECs are evaluated against the ATP, is illustrated in Figure 3. FFP method conditions (typically starting from mid-phase methods) are first developed and optimized, with robustness studies performed on the proposed method ECs.
A detailed risk assessment is then conducted, leading to two potential outcomes:
- The method meets the ATP with acceptable residual risk and no gaps in pertinent knowledge, making it ready for registrational validation.
- The method does not meet the ATP, has unacceptable residual risk, or has knowledge gaps.
From the RA, any gaps and risks are summarized and discussed. If necessary, any additional work to be performed is determined to reassess the method and better understand the gaps and/or risks. These studies will inform actions such as method changes or the implementation of appropriate controls to reduce the gaps and mitigate risks before another evaluation cycle.
Risk Assessment Tools
To simplify the evaluation during an analytical method RA, activities are divided into two broad categories: 1) sample preparation and 2) sample analysis, as shown in Figure 4, which provides some example subcategories for a typical LC assay. Specific subcategories and variables would be method type EC-dependent. Additionally, Ishikawa diagrams are used to group knowledge and collect information based on SME input, which is later transcribed into a spreadsheet with predefined questions. The 6 Ms (Mother Nature, Measurement, humanpower, Machine, Method, and Material) help visually cluster variables and knowledge. This approach keeps the discussion more focused and, through experience, reduces the time-consuming overlap between discussions of sample preparation variables and analysis variables. Using a spreadsheet format for the templates provides flexibility in collecting and editing information during the specific method RA. Spreadsheet templates with predefined lists of potential method concerns were collated for several common specific methods, including: 1) LC assay and impurities, 2) GC assay and impurities, 3) LC/ MS mutagenic impurities, and 4) LC In-Process-Controls (IPCs). These can be illustrative and used as starting points for any other type of method where an RA is warranted. The spreadsheets’ list of predefined areas of concern has also continued to evolve based on experiences and lessons learned.
Link to spreadsheet example
Risk Assessment Round 1 and tool use
During the initial RA (Round 1), the team identifies and assesses key (critical) parameters, potential hazards, gaps, and their impact on the ATP and the product’s critical quality attributes (CQAs) based on method knowledge and collective team experience. Maximizing the benefits from the RA depends on a combination of having the information collated and communicated clearly through the tools and templates, coupled with SME engagement. SMEs are essential, as they leverage their experience and insight when evaluating areas of potential risks due to gaps in knowledge or conditions with risks of failure. They inform decisions and direct the output by formulating next steps, such as performing further experiments, gathering knowledge [e.g., through method design of experiments (DoEs)], advising changes, or suggesting simple/robust controls.
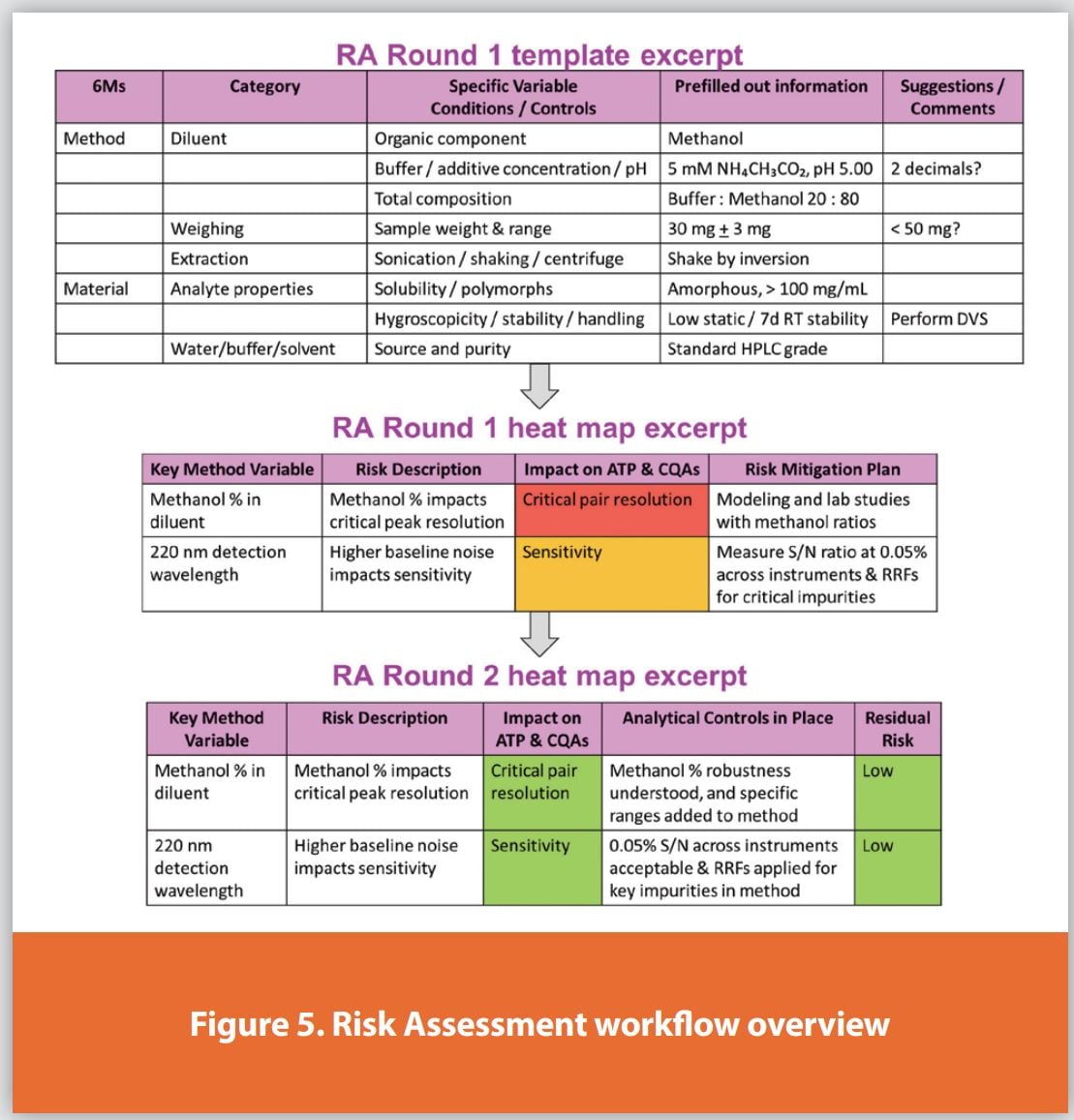
A cornerstone of the program is the pre-populated method assessment spreadsheet tool review. During the RA meeting, this pre-filled-out evaluation of the method’s variables and whether they meet the ATP or affect a CQA is reviewed line-by-line. A conceptual view of the tool is shown in Figure 5. In detail, the spreadsheet assessment tool has the following tabs and structure:
- An introduction tab capturing the project, scientists, analyte, timeline, and ATP information.
- A sample preparation assessment tab with columns for the 6 Ms, variables, specific concerns with background information, a column for inputting responses on the method under evaluation, and comments (usually suggesting next steps for activities, gaps, or related project experiences).
- A sample analysis technique-specific tab (e.g., chromatography) with columns for the 6 Ms, technique-centric variables, specific concerns with background information, a column for inputting responses on the method under evaluation, and comments.
- An RA Heat Map Round 1 tab, which collates only the determined risks or gaps, the concern, the grade of impact on ATP or CQA (red or yellow), and the mitigation plan determined by the team.
- An RA Heat Map Round 2 tab (discussed further in the next section), which contains the risk description, control strategy following implementation of changes or gathering further knowledge, and the revised grade of impact on ATP or CQA (green, yellow, or red).
Using the spreadsheet tool to lead the discussion, performance is compared to that expected, and any discussion with advice or next steps is captured in the comments section. If deemed a risk or gap by the team during Round 1, it will be graded as either medium (yellow) or high (red) risk. Note that, for simplicity, acceptable or no risk is not color-coded green. Any unusual method conditions, such as uncommon instrumentation, hard-to-source materials, atypical operating parameters, or parameters near the edge of typical calibrations, are evaluated in more detail. Where possible, data from the method innovator lab and from receiving labs can be shown to confirm the reproducibility.
Risk Assessment Round 2 and tool use
After implementing mitigation measures and/or gathering information to address knowledge gaps, the team performs a second round of RA. Where residual risks are deemed acceptable, the Heat Map Round 2 is updated accordingly with the changes captured and revised grade of impact on the ATP or CQA.
Examples of method controls to make residual risks acceptable might include:
- Adjusting unit operations to reduce error in sample preparation, such as increasing minimum weights or specifying specific materials.
- Verifying suitable alternate sources of reagents (e.g., buffers or solvents) or instrumental consumables (e.g., columns or liners).
- Confirming key method parameters through added System Suitability Tests and criteria to ensure control over method performance.
- Implementing more robust procedures, such as controlling column temperature within a tighter range, to reduce failures and enhance understanding for continual improvements or regulatory flexibility.
Throughout the assessment process, the RA SME team should share planning and experiences with method developers, implementers, and collaborators to gain a detailed understanding of the method development and control needs. If residual risks are not acceptable, the team should determine an alternative course of action, such as further method optimization using modeling software and additional evaluation of operating parameters.
Program Achievements and Growth
This program has supported more than eight projects and 40 methods within BMS over seven years. The identification of medium to high-risk areas across the portfolio of projects has led to the implementation of various method controls to mitigate risks, resulting in 24% minor method changes and 5% major method changes. The feedback from this program has contributed to improvements in method development through training, timely SME engagements, aligned expectations, and evolved method development workflows.
In Figure 6, the method performance risks identified over seven years and across many methods are plotted against the 6 Ms. In the analytical method development workflow, identifying and mitigating risks and knowledge gaps associated with the analytical method is a significant milestone. Among the 6 Ms, the M for Method area was by far the most significant source of risk, accounting for about 71% of medium to high risks. This is not entirely surprising, as this area has the greatest number of variables impacting the analytical methods, and most are also chosen specifically for the analyte of interest during development (e.g., sample diluent, chromatographic column type, and mobile phase composition). The specific risks associated with Materials included compounds of interest, contact materials (e.g., glassware), buffer and solvent sources, and other method-related chemicals used in the analysis process. Risks from critical Materials related to the methods accounted for 14% of the total risk. MMachine, which includes variables associated with GCs, LCs, pH meters, analytical balances, etc., was another vital component highlighted during the risk analysis process. Risks related to Machine accounted for about 10%. The other 3 Ms (Mother Nature, Measurement, and huManpower) only accounted for 5% of the risks.
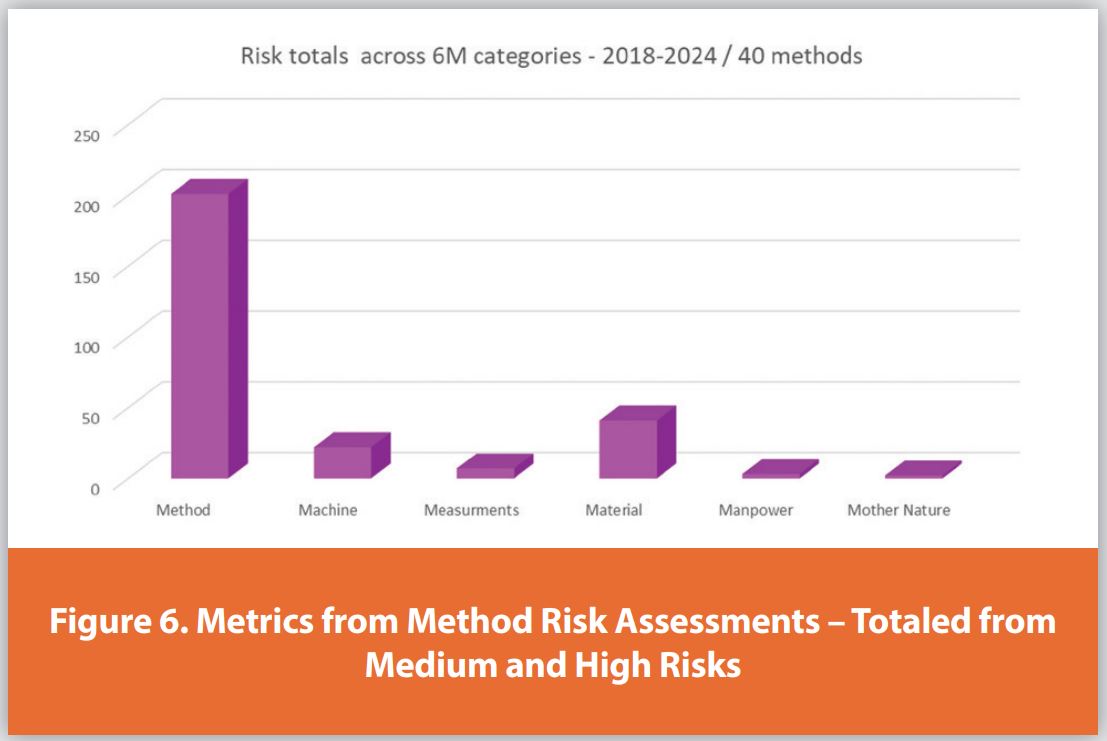
Communication of RA experiences also leads to greater awareness of common risks, enabling the identification of opportunities to implement method development strategies that reduce the likelihood of risk. However, this analysis also reveals that high and medium level concerns have not been eliminated, likely due to specific method conditions and/or analyte properties. This underscores the continued need for the RA program, as well as the continued refinement of the spreadsheet tools. Over the years, this program has accumulated a wealth of information and has significantly contributed to the identification of method risks and the improvement of many methods in BMS late-stage projects.
Conclusion
We have demonstrated a late-stage method development workflow that begins with an ATP and includes method development activities, 12 | | May/June 2025, and optimization to generate simple and robust established method conditions. This workflow integrates an RA tool applied to specific methods intended for commercial QC lab use. This program helps align expectations and working practices while gaining value from timely stakeholder involvement. Although only a small proportion of development projects are commercialized, this approach helps focus stage-appropriate eff orts where they have the most impact and return on investment. The RA delivers a comprehensive and clear evaluation through a stepwise approach, where the actionable Round 1 and Round 2 heat map tables drive method performance improvements and target mitigations against consistent expectations and criteria.
The program has been successfully applied to many methods over the last seven years, and the outcomes have been well-received by internal and external stakeholders. Additionally, its structure and detail have helped train and develop scientists in analytical development. The program continues to evolve as needed, incorporating recent ICH guidelines and benefiting from feedback from each iteration into the templated collection of variables and areas for potential concern. The mature guidance has empowered analytical chemists, enabled value through the use of stakeholders and SMEs at the appropriate time, provided consistent expectations with appropriate flexibility to meet specific project control needs, and embedded a simple-to-use tool that can be applied across the late-stage product portfolio.
The authors gratefully acknowledge the contributions from the BMS RA team: James Chadwick, Adrian Doggett, Brian He, Michelle Kubin, Robert Menger, Mallikarjun Narayanam, and Yan Zha.
References
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonized Tripartite Guideline. Pharmaceutical Development, Q8 (R2), 2009.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized Tripartite Guideline. Quality Risk Management, Q9, 2005.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized Tripartite Guideline. Pharmaceutical Quality System, Q10, 2008.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized Tripartite Guideline. Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities), Q11, 2012.
- L. Li, P. Tattersall, Q. Wang, J. Shackman, L. Ma, Doggett, A., A Practical Analytical Risk Survey Program for Active Pharmaceutical Ingredient Synthesis that Enhances Analytical Control Strategies for Early and Mid-Stage Development, American Pharmaceutical Review, 25 (13) 18.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized Tripartite Guideline. Validation of Analytical Procedures, Q2(R2), 2022.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized Tripartite Guideline. Analytical Procedure Development, Q14, 2022.
Author Details
Peter Tattersall, Lianjia Ma, Qinggang Wang, Li Li, and Jonathan Shackman- Chemical Process Development, Bristol Myers Squibb
Publication Details
This article appeared in American Pharmaceutical Review:Vol. 28, No. 4
May/June 2025Pages: 8-12Subscribe to our e-Newsletters.
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!