Drug Carriers and Difficult-to-Deliver Drugs
Traditional pharmaceuticals rarely demonstrate specific affinity towards the site of their action and as a rule, they distribute throughout the body upon administration. To reach the action site, a pharmaceutical agent has to overcome the inactivating action of the aggressive biological medium and cross a variety of biological barriers, which frequently results in at least partial drug inactivation/degradation and unfavorable pharmacokinetics and biodistribution. In addition, many pharmaceutical agents could provoke multiple undesirable sideeffects in normal organs, tissues and cells. To solve these complicating issues, various systems for drug delivery are suggested, and some of those even have already found their way to clinic.
In very general terms, these drug delivery systems are supposed to fulfill three major tasks:
- Protect a drug from the body (which is especially true for unstable pharmaceuticals, such as protein and peptide drugs);
- Protect the body from a drug (which is especially true for highly toxic pharmaceuticals, such as chemotherapeutic drugs); and
- Favorably change drug pharmacokinetics (which is especially required in cases when even stable and non-toxic drugs could be cleared from the body too rapidly to stably maintain their therapeutic concentration in the blood).
Currently, we have a broad set of pharmaceutical nanocarriers (liposomes, micelles, solid lipid nanoparticles, niosomes, dendrimers, polymeric nanoparticles, etc.) at our disposal, along with the means to load them with various drugs and adjust their surface properties to make them long-circulating, targeted, stimuli-sensitive, and even multifunctional.
Among numerous pharmaceuticals to be delivered in the body by means of various drug delivery systems, some are especially difficult to handle because of their intrinsic properties. Those could be unified under the general names of “non-deliverable” or “difficultto- deliver” pharmaceuticals. The following features make up pharmaceuticals that belong to this group:
1. Very poor solubility
Poor solubility could be benefi cial for drug activity, since it assists drug penetration into cells directly through cell membranes, but it also results in poor bioavailability and difficulty in maintaining drug therapeutic concentrations in the blood.
2. Very low biological stability
Many drugs, especially those of protein or peptide nature, are quickly destroyed by body enzymes. This is also true for siRNA-based pharmaceutical agents that are increasing in popularity, but are highly sensitive to nucleolytic degradation.
3. Very fast clearance from the blood
In such cases, the creation of the desired blood concentration is very difficult and frequently requires constant infusion.
4. A drug’s inability to reach its biological target
(e.g., to go where we want it to go). This is especially true for the cases when a drug should act on intracellular pathways or on individual intracellular organelles, such as lysosomes or mitochondria).
Here, we will briefly consider how to overcome some of those challenges with the help of nanosized drug delivery systems. Challenges one through three will be considered, since intracellular or individual organelle drug targeting is already a subject of multiple publications (see reference [1]).
Delivering poorly soluble pharmaceuticals
One of the most established approaches to increasing the bioavailability of poorly soluble pharmaceuticals is solubilization into polymeric micelles [2, 3].
Micelles solubilize insoluble drugs and increase their bioavailability. They can stay in body (in the blood) long enough to provide gradual accumulation in the required area, and their size permits them to accumulate in body regions with leaky vasculature via enhanced permeability and retention (EPR). They can become targeted by the attachment of specifi c molecules to their surface, and they can be prepared in large quantities easily and reproducibly. Being in a micellar form, the drug is well protected from the effects of biological surroundings and does not provoke undesirable side effects.
Micelles are amphiphilic colloidal dispersions with particle sizes within the 5 to 50-100 nm range that spontaneously form from amphiphilic substances, which are molecules consisting of two distinct fragments with opposite affinities towards a given solvent [4, 5]. The principal scheme of micelle formation from an amphiphilic molecule in an aqueous medium can be seen in Figure 1.
 Figure 1. Schematic of micelle formation and loading with the drug. Highly insoluble hydrophobic drug (5) goes into the micelle core; highly soluble drug (1) could adsorb into the micelle corona; drugs with intermediate solubilities (2-4) go to intermediate positions.
Figure 1. Schematic of micelle formation and loading with the drug. Highly insoluble hydrophobic drug (5) goes into the micelle core; highly soluble drug (1) could adsorb into the micelle corona; drugs with intermediate solubilities (2-4) go to intermediate positions.The major driving force behind this self-association is the decrease of free energy of the system due to removal of hydrophobic fragments from the aqueous surroundings, with the formation of micelle core stabilized with hydrophilic blocks exposed into water [6]. The concentration of a monomeric amphiphile at which micelles appear is called the critical micelle concentration (CMC). Mixed micelles could be composed of several amphiphiles, and the CMC of the mixture can be calculated from CMC values of individual components and their molar fraction (M) in the mixture, as follows:
1/CMC = M1/CMC1 + M2/CMC2
The most important property of micelles to consider is their ability to increase the solubility of poorly water-soluble substances (refer to Figure 1). In non-ionic micelles, water concentrations decrease from the surface towards the core of the micelle. As a result, in aqueous systems, nonpolar/water-insoluble molecules will be solubilized within the micelle core and will be well protected from any external factors. For pharmaceutical micelles, optimal CMC value should be in a low micromolar region, and the loading efficacy towards a hydrophobic drug should be between 5 and 25% wt.
In an absolute majority of cases, in polymeric amphiphiles, PEG blocks (the most frequently occuring hydrophilic blocks) have a molecular weight from 1 to 15 kDa and form micelle corona, while the length of a hydrophobic core-forming block is close or a bit lower than that of a hydrophilic block [7]. Though some other hydrophilic polymers may be used to make corona blocks [8, 9], PEG remains the hydrophilic block of choice. At the same time, a variety of polymers may be used to build hydrophobic, core-forming blocks including polypropylene oxide, poly-L-lysine, poly-aspartic acid, poly-β-benzoyl-L-aspartate, poly-γ-benzyl-L-glutamate, polycaprolactone, poly-D,L-lactic acid, and few others (see review in [10]). In some cases, phospholipid residues (short, yet extremely hydrophobic due to the presence of two longchain fatty acyl groups) can also be successfully used as hydrophobic core-forming groups [11].
Usually, it is accepted that micelles are spherical particles with clear distinction between core and corona compartments. However, it was shown that block copolymer aggregates can exist in many different morphologies, including spheres, rods, various vesicles, tubules, lamellae, hexagonally-packed hollow hoops, and various mixed and combined morphologies, depending on copolymer composition and concentration [12, 13].
A drug can be incorporated into micelles by simple physical entrapment or via preliminary covalent or electrostatic binding with a hydrophobic block of a micelle-forming amphiphilic block co-polymer. If a drug is attached to a hydrophobic block, its incorporation into the micelle core proceeds spontaneously and simultaneously with micelle formation. If a free drug should be physically entrapped into the micelle, various preparation methods are used, such as direct dissolution protocol (polymer solution in water is added to a drug dried from an organic solvent) or dialysis method (drug is dissolved together with a micelle-forming polymer in an organic solvent with further dialysis against water). Polymeric micelles effectively solubilize such drugs as paclitaxel, camptothecin, ceramide, curcumin, diazepam, indomethacin, adriamicin, anthracycline antibiotics, and polynucleotides [2]. Thus, successful solubilization of paclitaxel by micelle-forming co-polymers of PEG and poly(D,L-lactic acid) was described almost 20 years ago [12, 14, 15]. The use of micelles permitted an increase in paclitaxel solubility from less than 0.1 to 20 mg/ml. There are numerous examples of the successful use of poorly-soluble drugs in the micellized form, and paclitaxel in polymeric micelles (Genexol®) is approved for clinical use in some countries.
Three targeting mechanisms can be seen for micelles. The first one is based on micelle spontaneous penetration into the interstitium through the leaky vasculature (EPR effect). Thus, it was repeatedly shown that micelle-incorporated drugs (see, for example, [16]) accumulate much better in tumors than in not-target tissues. The second targeting mechanism is based on the fact that many pathological processes in various tissues and organs are accompanied with local temperature increase and/or acidosis, and micelles made of thermo- or pH-sensitive components, such as poly(N-isopropylacrylamide) and its co-polymers, can disintegrate in such areas, releasing the micelle-incorporated drug [5, 17]. By the third mechanism, specific ligands can be attached to the water-exposed termini of hydrophilic blocks, such as antibodies and/ or certain sugar moieties [18].
Still, the search for alternative approaches to delivering poorlysoluble drugs continues. In the last decade, electrostatic layer-by layer (LbL) self-assembly has been developed as a practical and versatile method for nanocoating and encapsulation [19-22]. A technique for layer-by-layer (LbL) self-assembly of thin films by means of alternate adsorption of oppositely-charged linear polyions was introduced in the mid 1990s [19, 20, 23]. The basis of the method involves resaturation of polyion adsorption, resulting in the reversal of the terminal surface charge of the film after deposition of each layer. The method provides the possibility of designing ultrathin multilayer films with a precision better than one nanometer, with defined molecular composition. The assembly process elaborated for planar solid supports was adapted for nano- and microtemplates (colloid particles with sizes of 30 nm to hundreds nanometers, e.g. nanoparticles, biological cells) [24- 27]. In this process, a polycation solution is added to a suspension of colloid particles and, after adsorption saturation, the particles are separated from free polycations in solution. Then, a polyanion layer is deposited. One can deposit any number of polyion layers on the shell by repeating the procedure. A typical thickness of the multilayer shell wall is 3-30 nm, depending on the number of polycation/polyanion pairs [25, 28, 29].
Polyions predominately used in the shell assembly include polycations (poly(ethyleneimine) (PEI), poly(dimethyldiallyl ammonium chloride) (PDDA), poly(allylamine) (PAH), polylysine (PLL), polyarginine (PLA), chitosan, dextran amine, protamine sulfate) and polyanions (poly(styrene sulfonate) (PSS), poly(acrylic acid), dextran sulfate, sodium alginate, gelatin, chondroitin sulfate, heparin, DNA), many of those being biocompatible and recognized as safe.
We have suggested using this approach for making stable nanocolloids of poorly soluble drugs and introduced a preliminary stage of powderizing drug crystals by ultrasound into nanoparticles prior to the LBL coating [30]; see Figure 2.
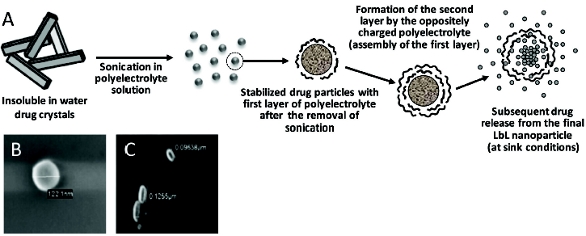 Figure 2. LBL colloids of poorly soluble drugs. (A) Schematics of combined ultrasound-LBL-mediated solubilization of poorly soluble drugs; (B) LBL nanoparticles of tamoxifen; (C) LBL nanoparticles of paclitaxel.
Figure 2. LBL colloids of poorly soluble drugs. (A) Schematics of combined ultrasound-LBL-mediated solubilization of poorly soluble drugs; (B) LBL nanoparticles of tamoxifen; (C) LBL nanoparticles of paclitaxel.Our initial experiments have been performed with the several poorlysoluble anti-cancer drugs including paclitaxel and tamoxifen, and with such polymers as positively charged poly(allylamine hydrochloride) (PAH), FITC-labeled PAH, and poly(dimethyldiallylamide ammonium chloride) (PDDA) and negatively charged sodium poly(styrene sulfonate) (PSS). The particle size of all the drug samples formulated by the LbL technology into nanocolloidal state was confi rmed by scanning electron microscopy and confocal fluorescent microscopy. Figure 2 demonstrates SEM images of prepared colloidal drug particles. Particles of tamoxifen demonstrate mainly spherical shape and have a diameter of ca. 120 nm. LbL-coated paclitaxel has the elongated rod-like shape with approximate measurements of 50 x 120 nm. Taking into account that the thickness of a single polymeric layer is approximately 1.5 nm, we can easily calculate that the drug content in stable nanocolloidal particles of poorly soluble drugs ranges from 85 to 95% wt. (in the case of a single layer coating), which is dramatically higher than any other solubilization method. Using variable number of bilayers will allow both for the quantity of the loaded drug and drug release rate from nanoparticles. Naturally, the release slows down as the number of polyelectrolyte layers in the shell increases. At sink conditions, non-coated tamoxifen crystals (both without and with sonication) solubilized within approximately 2 hours, while LbL coating easily extends this time to approximately 10 hours. LbL-coated drug nanoparticles can also be derivatized on the surface in order to impart the various additional properties including targetability and/ or longevity.
Delivering unstable pharmaceuticals
An interesting example to be considered here is siRNA, which is gaining an increased recognition as a potential drug with a broad variety of applications. However, the real practical use of siRNA for clinical purposes is severely hindered by its very low stability in biological surroundings and the lack of simple and efficient delivery systems for siRNA [31, 32]. Naked siRNA degrades quickly and possesses poor cellular uptake. Further, high doses and repeated administration are often necessary for activity. siRNA delivery systems should be properly designed to improve the stability of siRNA upon in vivo administration, deliver siRNA specifi cally to the target tissue site, and facilitate the cellular uptake of siRNA into target cells [33, 34]. To address these problems, several kinds of nano-sized cationic delivery systems have been studied, which are based on polyelectrolyte complexes resulting from the electrostatic interaction between the negatively charged siRNA and cationic polymers or lipids [35]. However, cationic agents used to form polyplexes with the siRNA, such as polyethyleneimine, often show strong toxicity, thus limiting their clinical application [35]. It has also been shown that the direct conjugation of aptamers, lipids, peptides, proteins, or polymers to siRNA could improve its in vivo pharmacokinetics, increase half-life, and increase delivery effi ciency [36-39]. However, chemical modifi cation of siRNA can affect its activity and specifi city, and in such conjugates, siRNA still remains open for degradation by nucleases.
We have attempted to prepare a nanoparticle-based delivery system for siRNA, in which siRNA remains protected from the degradation on its route to the target by making the reversible (reducible) conjugates of siRNA with phospholipid (L-S-S-siRNA) and incorporating those in polymeric micelles made of polyethylene glycol-phosphatidyl ethanolamine (PEG-PE) conjugates [40]. We have developed synthetic schemes to prepare L-siRNA conjugates with the reducible –S-S-bond between lipid and siRNA moieties (to liberate free siRNA from micelles after their delivery inside cells, where high glutathione creates reducing conditions); see Figure 3.
 Figure 2. LBL colloids of poorly soluble drugs. (A) Schematics of combined ultrasound-LBL-mediated solubilization of poorly soluble drugs; (B) LBL nanoparticles of tamoxifen; (C) LBL nanoparticles of paclitaxel.
Figure 2. LBL colloids of poorly soluble drugs. (A) Schematics of combined ultrasound-LBL-mediated solubilization of poorly soluble drugs; (B) LBL nanoparticles of tamoxifen; (C) LBL nanoparticles of paclitaxel.L-siRNA forms stable mixed micelles with PEG-PE, the CMC value being ca. 6x10-6M and the size of about 10 nm with narrow size distribution. We have also shown that when incubated in the presence of 10 mM glutathione (the concentration that could be found inside cells), free non-modified siRNA releases from such mixed micelles due to the reduction of the –S-S-bond. Importantly, being a component of mixed micelles, siRNA is well protected against degradation by nucleases and remains intact for at least 24 hours in the presence of nucleases, which completely degrade free siRNA within less than 30 minutes (Figure 3).
Such stabilization can be explained by steric hindrances created by the micelle surface for an enzymatic attack. Our preliminary experiments on the silencing property of micellar siRNA demonstrated that, in addition to intracellular uptake, L-siRNA/PEG-PE micelles were able to provoke a significant down-regulation of the target gene and decrease the GFP production in pGFP-transfected C166-GFP endothelial cells, thus confirming the release of free, undamaged siRNA from the mixed micelles inside cells.
Overcoming Fast Clearance
Here, we will consider an example relating to the use of imaging agents. Computed Tomography (CT) represents an imaging modality with high spatial and temporal resolution. The diagnostic value of CT might be further increased when contrast agents (substances containing X-ray absorbing heavy elements, such as iodine) are used to attenuate tissues and organs of interest. Because providing diagnostically acceptable imaging requires the iodine concentration on the order of millimoles per ml of tissue [41], large doses of low-molecular-weight CT contrast agent, such as iodine-containing organic molecules, are normally administered to patients. The selective enhancement of blood upon such administration is brief due to rapid extravasation and clearance. At the same time, blood pool imaging is of special interest for the evaluation of the current state of blood flow and for the discovery of irregularities caused by atherosclerotic lesions, thrombi or tumors. In order to serve as a nanoparticulate CT contrast agent whose distribution is limited to the blood pool, the nanoparticulate has to be of a size larger than fenestrated capillaries (>10 nm), have resistance to phagocytosis, and have the radiopaque moiety structurally incorporated within the particulate.
With this in mind, we have suggested a micelle-forming block-copolymer of methoxy polyethylene glycol and iodine-substituted poly-L-lysine [42, 43]. This copolymer easily micellizes within solution, forming stable and heavily iodine-loaded particles (up to 35% of iodine by weight) with a size well below 100 nm. The formation of a polymeric micelle with the iodine-containing core and hydrophilic corona consisting of PEG chain is schematically shown on Figure 4.
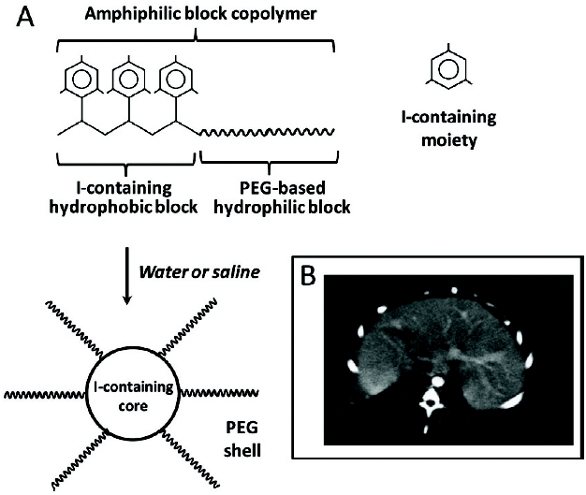 Figure 4. Long-circulating CT contrast for blood pool imaging. (A) Schematics of preparation of iodine-containing longcirculating micelles; (B) Liver blood pool imaging in rabbit 60 min after contrast administration.
Figure 4. Long-circulating CT contrast for blood pool imaging. (A) Schematics of preparation of iodine-containing longcirculating micelles; (B) Liver blood pool imaging in rabbit 60 min after contrast administration.In vivo, the iodine-containing polymeric micelles demonstrate prolonged circulation time and upon their intravenous administration in rabbits, a signifi cant enhancement of 3 to 4-fold in the blood pool (aorta and heart) was visually observed for at least a period of 2 hours following the injection (Figure 4). Thus, the use of nanopreparation resulted in the contrast agent with properties not provided by the parent contrast.
The examples presented clearly show significant potential of nanotechnological approaches in resolving many issues hindering the practical application of difficult-to-deliver pharmaceutical agents.
Author biography
Vladimir P. Torchilin, Ph.D., is a University Distinguished Professor and Director, Center for Pharmaceutical Biotechnology and Nanomedicine, Northeastern University, Boston. He has published more than 500 original papers, reviews and book chapters, wrote and edited 10 books and holds more than 40 patents. He is Edltor-in-Chief of Cunent Drug Discovery Technologies and of Drug Delivery. Professor Torchilin won numerous national and international awards and served as a President of the CRS in 2005-2006. Times Higher Education ranked him number 2 among top world scientists in pharmacology for 2001-2010.
References
- Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng 2006; 8: 343-75.
- Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res 2007; 24: 1-16.
- Sawant RR, Jhaveri AM, Torchilin VP. Immunomicelles for advancing personalized therapy. Adv Drug Deliv Rev 2012; 64: 1436-46.
- Mittal KL, Lindman B, editors. Surfactants in Solution. New York: Plenum Press; 1991.
- Jones M, Leroux J. Polymeric micelles - a new generation of colloidal drug carriers. Eur J Pharm Biopharm 1999; 48: 101-11.
- Martin A, editor. Physical Pharmacy. Philadelphia, PA: Lippinkott Williams and Wilkins; 1993.
- Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T. Thermo-responsive polymer nanoparticles with a core-shell micelle structure as site-specifi c drug carriers. J Control Release 1997; 48: 157-64.
- Torchilin VP, Shtilman MI, Trubetskoy VS, Whiteman K, Milstein AM. Amphiphilic vinyl polymers effectively prolong liposome circulation time in vivo. Biochim Biophys Acta 1994; 1195: 181-4.
- Torchilin VP, Trubetskoy VS, Whiteman KR, Caliceti P, Ferruti P, Veronese FM. New synthetic amphiphilic polymers for steric protection of liposomes in vivo. J Pharm Sci 1995; 84: 1049-53.
- Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release 2001; 73: 137-72.
- Trubetskoy VS, Torchilin, V.P. Use of polyoxyethylene-lipid conjugates as long-circulating carriers for delivery of therapeutic and diagnostoc agents. Adv Drug Deliv Rev 1995; 16: 311-20.
- Zhang L, Eisenberg A. Multiple morphologies and haracteristics of “crew-cut” micelle-like aggregates of polystyrene-b-poly(acrylic acid) diblock copolymers in aqueous solutions. J Am Chem Soc 1996; 118: 3168-72.
- Allen C, Maysinger D, Eisenberg A. Nano-engeneering block copolymer aggregates for drug delivery. Coll Surf B: Biointerf 1999; 16: 1-35.
- Zhang L, Bartels C, Yu J, Shen H, Eisenberg A. Mesosized crystal-like structure of hexagonally packed hollow hoops by solution self-assembly of diblock copolymers Phys Rev Lett 1997; 79: 5034-7.
- Ramaswamy M, Zhang X, Burt HM, Wasan KM. Human plasma distribution of free paclitaxel and paclitaxel associated with diblock copolymers. J Pharm Sci 1997; 86: 460-4.
- Kwon G, Suwa S, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly(ethylene oxide-aspartate) block copolymeradriamycin conjugates. J Control Release 1994; 29: 17-23.
- Kohori F, Sakai K, Aoyagi T, Yokoyama M, Sakurai Y, Okano T. Preparation and characterization of thermally responsive block copolymer micelles comprising poly(N-isopropylacrylamide-b-DLlactide). J Control Release 1998; 55: 87-98.
- Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci U S A 2003; 100: 6039-44.
- Lvov Y, Ariga K, Ichinose I, Kunitake T. Assembly of multicomponent protein fi lms by means of electrostatic layer-by-layer absorption. J Am Chem Soc 1995; 117: 6117-22.
- Lvov Y, Lu Z, Schenkman J, Rusling J. Direct electrochemistry of myoglobin and cytochrome P450 in alternate layer-by-layer fi lms with DNA. J Am Chem Soc 1998; 120: 4073-80
- Yoo Y, Shiratori S, Rubner M. Controlling bilayer composition and surface wettability of sequentially adsorbed multilayers of weak polyelectrolytes. Macromolecules 1998; 31: 4309-18.
- Mendelson J, Barret C, Chan V, Pal A, Mayes A, Rubner M. Fabrication of microporous thin fi lms from polyelectrolyte multilayers. Langmuir 2000; 16: 5017-23.
- Decher G. Fuzzy nanoassemblies:toward layered polymeric multicomposites. Science 1997; 227: 1232-7.
- Donath E, Sukhorukov GB, Caruso F, Davis H. Novel hollow polymer shells by colloid-templated assembly of polyelectrolytes. Angew Chem Int Ed Engl 1998; 37: 22202-2205.
- Lvov Y, Antipov A, Mohwald H, Sukhorukov GB. Urease encapsulation in nanoorganized microshells. Nano Lett 2001; 1: 125-8.
- 26. Lu Z, Prouty MD, Guo Z, Golub VO, Kumar CS, Lvov YM. Magnetic switch of permeability for polyelectrolyte microcapsules embedded with Co@Au nanoparticles. Langmuir 2005; 21: 2042-50.
- Shutava TG, Kommireddy DS, Lvov YM. Layer-by-layer enzyme/polyelectrolyte fi lms as a functional protective barrier in oxidizing media. J Am Chem Soc 2006; 128: 9926-34.
- Caruso F, Caruso RA, Mohwald H. Nanoengineering of inorganic and hybrid hollow spheres by colloidal templating. Science 1998; 282: 1111-4.
- Lvov Y, Caruso F. Biocolloids with ordered urease multilayer shells as enzymatic reactors. Anal Chem 2001; 73: 4212-7.
- Agarwal A, Lvov Y, Sawant R, Torchilin V. Stable nanocolloids of poorly soluble drugs with high drug content prepared using the combination of sonication and layer-by-layer technology. J Control Release 2008; 128: 255-60.
- Jeong JH, Kim SW, Park TG. Molecular design of functional polymers for gene therapy Prog Polym Sci 2007; 32: 1239-74.
- Jeong JH, Mok H, Oh YK, Park TG. siRNA conjugate delivery systems. Bioconjug Chemistry 2009; 20: 5-14.
- De Paula D, Bentley MV, Mahato RI. Hydrophobization and bioconjugation for enhanced siRNA delivery and targeting. RNA 2007; 13: 431-56.Dorsett Y, Tuschl T. siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov 2004; 3: 318-29.
- Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release 2007; 121: 64-73.
- Lorenz C, Hadwiger P, John M, Vornlocher HP, Unverzagt C. Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells. Bioorg Med Chem Lett 2004; 14: 4975-7.
- Moschos SA, Jones SW, Perry MM, et al. Lung delivery studies using siRNA conjugated to TAT(48-60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem 2007; 18: 1450-9.
- Nishina K, Unno T, Uno Y, et al. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol Ther 2008; 16: 734-40.
- Soutschek J, Akinc A, Bramlage B, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004; 432: 173-8.
- Musacchio T, Vaze O, D’Souza G, Torchilin VP. Effective stabilization and delivery of siRNA: reversible siRNA-phospholipid conjugate in nanosized mixed polymeric micelles. Bioconjug Chem 2010; 21: 1530-6.
- Wolf GL. Targeted delivery of imaging agents: an overview. In: Torchilin VP, editor. Handbook of targeted delivery of imaging agents. Boca Raton: CRC Press; 1995. p. 3-22.
- Trubetskoy VS, Gazelle GS, Wolf GL, Torchilin VP. Block-copolymer of polyethylene glycol and polylysine as a carrier of organic iodine: design of long-circulating particulate contrast medium for X-ray computed tomography. J Drug Target 1997; 4: 381-8.
- Torchilin VP, Frank-Kamenetsky MD, Wolf GL. CT visualization of blood pool in rats by using long-circulating, iodine-containing micelles. Acad Radiol 1999; 6: 61-5.