Abstract
Supercritical fluid chromatography (SFC) with electron capture detection (ECD) was evaluated for use in determination of low levels of alkylating agents. Several alkylating agents, varying widely in chemical functionality, two sulfonates, two neutral nitrogen heterocycles, three organic amine salts, and five halocarbons were evaluated by packed column SFC for separation and detection with both pure CO2 and methanol-modified CO2. Successfully separated and detected analytes were evaluated for their determination from, or off of, an active pharmaceutical matrix, to mimic a lab like application. In general, the separation, detection, and detection limits varied for each compound type evaluated. Specifically, the amine salts failed to elute regardless of the chromatographic protocol due to their insolubility in the CO2-based mobile phases. The nitrogen heterocycles required methanol-modified CO2, but they both gave relatively strong ECD signals. Halocarbons were readily chromatographed with pure CO2 after liquid-solid extraction from a spiked drug matrix. Electron capture detection limits in methanol-modified CO2 for both 3, 6-dichloropyridazine and 3-chloro-6-chloromethylpyridine were 0.1μg/mL.
Introduction
Alkylating agents are used in the synthesis of many active pharmaceutical ingredients. The possibility of these agents being carried through into the final product coupled with their inherent genotoxic activity require vigorous analytical testing and monitoring. However, due to the low levels allowed and the chemical properties associated with different classes of alkylating agents, this analytical monitoring poses a unique challenge.
A proposal and rationale for setting limits on the presence of these agents has been documented [1], however analytical testing is still performed on a case by case basis. In general, the low level at which these agents are to be controlled at (typically ppm level or lower), requires a sensitivity that typical HPLC/UV analysis does not provide, while the fragile thermal stability of many of these reactive agents precludes the use of gas chromatography (GC) with FID or ECD in their analysis. This combination of alkylating agent’s attributes points towards the use of SFC with GC type detectors for their determination.
SFC fills the gap between GC and liquid chromatography (LC) in a variety of ways, and while its liquid like solvating power, its gas like diffusivity, and environmentally and economically preferable alternative to conventional liquid mobile phases are usually highlighted, it is the ability to interface to both GC-like and LClike detectors and its adaptability of all column dimensions to the supercritical fluid that uniquely expands SFC’s functionality.
Although first demonstrated in 1962, SFC only became seriously considered as a separation technique in the 1980’s. Initially, the primary thrust was placed on open tubular columns and pure carbon dioxide rather than on packed columns with modified carbon dioxide mobile phases as is the case today [2]. Under the earlier conditions popular flame-based detectors were routinely employed [3]. Traditional mass spectrometric detection was also easily implemented. The current SFC landscape has changed dramatically with packed columns and modified, compressible mobile phases being used to solve numerous separation problems involving a wide variety of chemical classes. In this regard, the use of flame-based detectors becomes more problematic because of the incorporation of modifier which is commonly, if not always associated with packed column SFC with polar analytes. This point is highlighted as, although previous positive results detection with capillary column SFC with FID detection have been reported [4, 5, 6, 7, 8], most instances are with non polar analytes which can elute with 100% CO2, and are in stark contrast to most pharmaceutical type analytes.
This manuscript attempts to assess the potential of modern day SFC with microbore packed columns for electron capture detection of several pharmaceutical alkylating agents. Although carbon dioxide and some modifiers are electron capturers, sensitive detection is still possible in the presence of certain modifiers. By altering the ECD temperature, the detectability of the ECD can be enhanced. A brief literature review regarding SFC-ECD will illustrate this point.
One of the first reports concerning SFC-ECD described the quantification of mefloquine (e.g. 2-piperidyl-2,8(trifluoromethyl)- 4-quinolinemethanol) in 0.1mL blood samples [9]. The SFC conditions included a silica gel packed- glass-lined steel column and a mobile phase of 0.15% n-butylamine and 1.0% methanol in supercritical n-pentane. The method had a detection limit of 7.5ng/mL for this analyte and the method exhibited good linearity and precision. Two years later SFC/ECD was employed to determine polychlorinated biphenyls (PCBs) in the environment [10]. The separation was conducted using an octadecyl-silica column and pure carbon dioxide as the mobile phase. The detection temperature of the ECD was set at 325°C. Base-line drift of the chromatogram was accompanied by an increase in carbon dioxide pressure which could be compensated for by an integrator.
A Ni63 ECD placed after a wall coated open tubular SFC column without any modification has also been reported [11]. On-column detection limits for nitro and halogenated compounds were found to be picograms and high femtograms respectively. Detector linear range was three to four orders of magnitude for the studied compounds. Good detector sensitivities were obtained for a number of thermally labile pesticides using SFC-ECD.
SFC-ECD was later applied to the analysis of felodipine, a drug indicated for the treatment of hypertension [12]. In this study, felodipine and its potential oxidative degradation product could be separated by SFC in under six minutes, thus increasing sample throughput 60% versus liquid chromatographic analysis.
Later five sulfonylurea herbicides and their breakdown products were on-line extracted from large water samples and separated by SFC on both a 25cm diol column (for rapid analysis) and a 1.6m column (for high resolution) using three different types of detectors simultaneously (e.g. UV, NPD, and ECD) [13]. The longer column was achieved with eight, the first a C18 column and the rest Hypersil silica, standard LC columns. Gradient delivery of methanol to the carbon dioxide from 1% to 16% was programmed with constant flow of 2mL/min at 60°C. Detection limits as low as 50 parts per trillion were observed. More recently, response surfaces were obtained for packed column SFC-ECD under various conditions in order to optimize the ECD for use with modified carbon dioxide [14, 15]. Limits of detection, correlation coefficients, and linear dynamic ranges were found to vary with increasing amounts of modifier for several nitrogen-containing and halogenated compounds. Low detection limits (pg) were achieved in the presence of 5% methanol-modified carbon dioxide. Applications directed toward the separation of compounds extracted from propellants and to simple phenylurea herbicides were presented.
In this brief report, we wish to illustrate the applicability of packed column SFC with methanol-modified CO2 and detection by ECD.
Experimental
Instrumentation
All separations were performed using either a syringe pump previously modified to be used with FID and ECD or an SFC equipped with an UV diode array detector. For the former system, a GC oven was used for packed column analysis with both FID and ECD detectors. Cyanopropyl, diol, and C18 (200 x 1.0mm, 5μm dp) stationary phases were used. Solutions for injection of each analyte were made up in hexane, methanol, or acetonitrile at concentrations ranging from 0.1ng/μL to 500ng/μL. An injection valve with an internal loop volume of 0.5μL was used for all packed column separations.
Packed Column SFC/ECD
The column flow rate was set to 40mL/min of expanded CO2 gas at 100atm. A tapered fixed restrictor was used for these separations. In this case, all samples were injected directly into the column without any split. The column outlet, however, was split such that 35mL/min of expanded CO2 gas was used with the FID; while only 5mL/min of expanded CO2 gas was used with ECD. Both detectors were used simultaneously. The FID auxiliary gas flow rates were set to 50mL/min of H2 and 400mL/min of air. For ECD, N2 was used as a make-up gas and the total flow was set to 60mL/ min. The following chromatographic conditions were used initially in this study:
Pressure: 80atm hold 5min 80 to 400atm in 25min at 12.8atm/min 400atm hold 5min
Oven Temperature: 80°C
Injection Volume: 0.5μL
Column: Cyanopropyl (200 x 1.0mm i.d., 5μm dp) or C18 (100 x 1.0mm i.d., 5μm dp)
The feasibility of using a high concentration of methanol as a CO2 modifier for detection of eluted analytes via ECD was also investigated. The modifier concentration ranged from 15 to 20%. Modifier was added to the mobile phase via a tee and mixing chamber using a secondary pump. For experiments with addition of modifier, all separations were performed at constant pressure. Solutions of each analyte were made up at concentrations ranging from 0.1ng/μL to 100ng/μL.
For all SFC separations, the CO2 pump was operated in flow mode rather than pressure mode. Methanol was mixed via an HPLC pump with CO2 and the resulting mixture was equilibrated in a mixing chamber which was filled with stainless steel balls. Modified CO2 was passed through the injection valve into the column which was located inside the oven. The outlet of the column was mixed with 300μL/min of methanol which was delivered from a second HPLC pump. Methanol containing either formic acid or ammonium acetate at either 0.1% or 1% for formic acid and 0.001% or 0.025% for ammonium acetate was used to aid the ionization of compounds in the spectrometer. In order to keep the column outlet pressure at 120atm, a 50μm capillary tube was used to restrict the flow of fluid into the MS. A fixed volume of mobile phase (CO2 modified with methanol and additive) entered the MS (approximately 1/3 of the total flow); while the remaining flow passed first through the UV detector flow cell then into a backpressure regulator which was maintained at 120atm.
Sample Preparation for Spiked Drug Matrix Study
Prior to SFC/ECD analysis of iodohexane and iodomethane from a drug matrix, 100 mg of a drug matrix was spiked with 1, 2, 5, and 10μg of the two analytes dissolved in hexane. Two extraction methods were used to obtain recovery of iodohexane or iodomethane from the spiked matrix. In the first method, 2mL of hexane was added directly to the solid spiked matrix which was agitated and centrifuged for 5 minutes at 5000 rpm. The hexane supernate was separated from the insoluble matrix and analyzed via SFC/ECD. In the second method, the entire spiked matrix was dissolved in 4mL of methanol. Then, 2mL of hexane was added to the methanol solution followed by the addition of 0.2mL of H2O. The resulting solution was shaken for 3-5 minutes, after which analysis of the upper hexane extract layer was pursued.
Results and Discussion
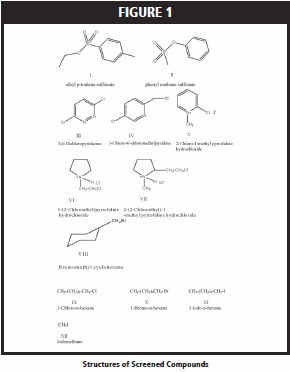
The purpose of this research was to study the elution of different potential alkylating agents from packed columns using carbon dioxide-based mobile phases with electron capture (ECD) detection. Various stationary phases were examined in conjunction with a mobile phase of either 100% CO2 or methanol-modified CO2. Figure 1 shows some of the alkylating agents, with accompanying structures that were used in this study. Each of these analytes presents unique chromatographic issues in terms of solubility, elution, and detection which must be addressed in order to achieve a successful separation.
Electron Capture Detection
Small diameter (1.0 mm) packed columns were used in this study as their lower flow rates versus analytical scale columns, should be beneficial with flame-based detectors. Initially, two stationary phases (cyano and diol) were tested for elution of the two sulfonates, 3,6-dichloropyridazine, and the neutral pyridine analyte, 3-chloro-6-chloromethylpyridine. The diol phase proved either to be too active with 100% CO2 as the mobile phase or pure CO2 had insufficient solvating power to dissolve the four analytes; thus none of the analytes tested could be successfully eluted. The results with the cyano phase and all analytes, were more promising as only the three amine salts (2-chloro-1-methyl pyridinium iodide, 1-(2-chloroethyl)pyrrolidine hydrochloride, and 2-(2-chloroethyl)-1- methyl pyrrolidine chloride) failed to elute with 100% CO2. This result was expected since CO2 has marginal solvating strength for ionic materials. As explained in the Experimental section, the outlet flow of the column (40mL/min of expanded CO2 gas) was split between the FID (35mL/min) and ECD (5mL/min). This was done to maintain detector stability, as a higher flow rate of CO2 into each detector caused more baseline noise, but results in the ECD monitoring only about 10% of the injected mass.
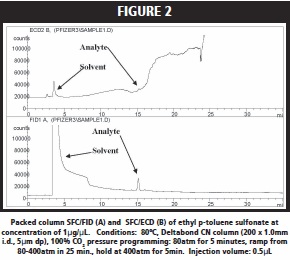
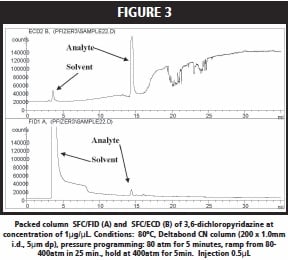
Figure 2 shows the chromatographic trace for the elution of ethyl p-toluene sulfonate with 100% CO2. A very weak peak was observed employing FID (injected mass ~ 100ng) with retention time of 15 minutes which was believed to be the sulfonate, Figure 2A. ECD (injected mass ~ 0.6ng) did not yield a good response for this analyte since it contained no halogen, Figure 2B. Next, phenyl methylsulfonate was injected onto the cyano packed column. A small peak with retention time of 14.5min in the FID (injected mass ~ 200ng) trace was observed, but the ECD (injected mass ~ 1.2ng) again did not show a strong response. On the other hand, 3, 6-dichloropyridazine showed a strong ECD (injected mass ~0.6ng) response for 100% CO2 elution of this analyte; while FID (injected mass ~ 150ng) showed a weak response, Figure 3. A similarly strong ECD response (injected mass ~ 6ng) was found for 3-chloro-5- chloromethylpyridine; while FID (injected mass ~ 200ng) again gave a poor response.
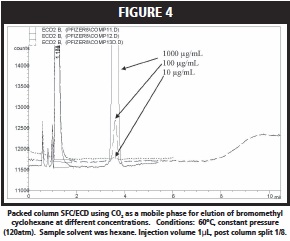
On the cyano column with CO2 at low pressure as the mobile phase, none of the halo alkanes were retained by the stationary phase. On the other hand, a C18 stationary phase proved to be better suited for elution of the haloalkanes. Figure 4 shows the chromatographic ECD trace for the elution of bromomethylcyclohexane dissolved in hexane at different concentrations (e.g. 1000, 100, and 10ng/μL). ECD did not show a strong response for this analyte at or below 10μg/mL, which was unexpected based on its being halogenated. The four additional halo compounds exhibited strong ECD signals, as iodohexane and iodomethane gave relatively stronger ECD responses compared to 1-chlorohexane and 1-bromohexane.
Through these experiments, acceptable (>0.1ng) MDQ values via ECD were obtained for 3, 6-dichloropyridazine, 3-chloro- 6-chloromethylpyridine, iodomethane, and 1-iodohexane (MDQ>0.1ng). Table I shows for comparison the measured detection limit of various compounds using the cyano column and 100% CO2 via FID and ECD. It should be noted that up to this point in the study, all separations were performed under isobaric conditions at relatively low pressures. For example, in order to obtain separation of the iodomethane peak from the small peaks arising from ECD responsive impurities in the injection solvent (hexane), a pressure of only 80atm was utilized.

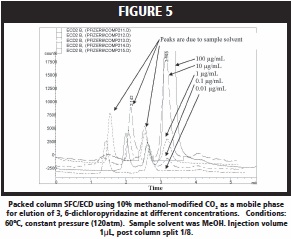
In order to fully evaluate SFC/ECD, and to expand its potential for elution and separation, the effect of methanol as a modifier on ECD response was studied. An increase in modifier concentration lowered the response of the ECD, but results showed that modified content up to 10% had a minimal effect on the detection limit of the analytes that were studied here. The oven temperature was reduced in this experiment which caused less bleed from the column thus reducing noise. Figure 5, for example, shows SFC/ECD traces using 10% methanol-modified CO2 as a mobile phase for elution of 3, 6-dichloropyridazine at various concentration (e.g. 100, 10, 1, 0.1, and 0.01ng/μL). ECD showed a strong response down to 0.1μg/ mL. Similar chromatographic behavior was observed for 3-chloro-5- chloromethylpyridine dissolved in methanol with a detection limit also near 0.1μg/mL. Alternatively when the analyte was dissolved in CH3CN, the detection limit was 1μg/mL.
SFC/ECD Analysis of Iodomethane and Iodohexane in a Drug Matrix
The next phase of study concerned the analysis of selected alkylating agents in a drug matrix. These experiments attempt to mimic laboratory situations where detection of a low level alkating agent must be done on an existing active pharmaceutical product. To do this, different concentrations (1, 2, 5, 10μg) of iodomethane and 1-iodohexane were spiked into a generic drug matrix, 100mg of a polar zwitterion with an arginine salt. The halocarbons were next extracted using two liquid-solid extraction methods prior to analysis via SFC/ECD. The solid matrix was extracted with hexane directly (DIR) or the matrix was dissolved in methanol and the resulting solution was extracted with hexane (DISO). Each sample was prepared in triplicate. Table II shows the ECD response to iodomethane at four concentrations using the two different extraction methods. As these results show, specific attention needs to be paid to the method of extraction of potential analytes from active pharmaceutical matrixes. In this case, in the method where the spiked matrix was dissolved in methanol followed by hexane extraction, there is loss of analyte. Various experiments in which different amounts of water were added in order to aid the partitioning of iodomethane into the hexane did not yield the expected improvement in recovery. This may point to a reactivity issue which may not be able to be solved with further exactions.

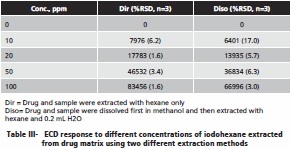
Similar experiments were performed for the analysis of 1-iodohexane. Table III shows the ECD response to iodohexane at four concentrations using the two different extraction methods. The iodohexane also showed a drop in extraction efficiency in going from direct extraction with hexane to the extraction where the matrix is first dissolved in methanol, but these losses are lower than seen with iodomethane. While recoveries are not quantitative, they are acceptable for trace level determination purposes. A plot of these recoveries showed good linearity.
Conclusions
Determination of low levels of alkylating agents must be assessed on a compound by compound basis because with their varying structures, no one technique or detection method is likely to be found as a universal tool. SFC was investigated specifically to bridge the gap between HPLC and GC through the use of HPLC type separations with GC type detectors, however not all analytes of interest were amenable to separation and or detection by SFC/ECD. The work illustrated the use of ECD as a detection method for some of these low level determinations. Lastly, experiments with analytes spiked onto an active pharmaceutical matrix provided good results and the indication that validation as a quantitative method or limits test may be achievable. In summary, this work illustrates the potential utility of SFC with ECD as a tool for low level determination of some alkylating agents in active pharmaceutical ingredients.
References
1. Muller L, et al, 2006, Regulatory Toxicology and Pharmacology, 44, 198-211.
2. Taylor LT, Chang CK, (1990) J. Chromatogr. Sci. 28, 357-366.
3. Chang CK, Taylor LT (1990) J. Chromatogr. Sci. 28, 29-33.
4. Mount DL, Patchen LC, Churchill FC (1990) J. Chromatogr. 527, 51-58.
5. Cecilia MH, Lancas FM (2000) J. High Resol. Chromatogr. 23, 7/8, 515-518
6. Bruheim I, Fooladi E, Lundanes E, Greibrokk T, (2001) J. Microcolumn Separations, 13(4) 156-162.
7. France JE, Snyder JM, King JW (1991) J. Chromatogr. 540, 271-278.
8. Di Sanzo FP, Yoder RE (1991) J. Chromatogr. Sci. 29, 4-7.
9. MacKay GA, Reed GD (1991) J. High Resol. Chromatogr. 14, 537-541.
10. Takashi Y, Horimoto Y, Yamada J, Nomura A (1992) Anal. Sciences 8, 811-815.
11. Later DW, Bornhop DJ, Lee ED, Henion JD, Weiboldt RC (1987) LC-GC, 5, 804-808.
12. Strode JTB, Taylor LT, Howard AH, Ip D, Brooks MA (1994) J. Pharm. Biomed. Anal., 12, 1003-1014.
13. Berger TA (1995) Chromatographia 41, 133-140.
14. Strode JTB, Taylor LT (1996) J. Chromatogr. 723, 361-69.
15. Berger T (1995) Chromatographia 41, 471-484
Mehdi Ashraf-Khorassani received his B.S. in chemistry from Jacksonville State University (Jacksonville, AL) in 1982, M.S. in Electro-analytical chemistry from University of Southwestern Louisiana (Lafayette, LA) in 1984, and Ph.D. in Analytical Chemistry (Chromatography) from Virginia Tech (Blacksburg, VA) in 1988. He started his carrier as a research scientist at Suprex Corporation in Pittsburgh, PA (1989-1992). From early 1992 till mid 1994 he joined Trabriat Modaress University (Tehran, Iran) as an assistant professor. In 1994 he joined Virginia Tech as a Senior Research Scientist in the Chemistry Department. His background is focused on various types of chromatography hyphenated to different detectors and advanced sample preparation techniques. During the past 20 years he has been involved with various analytical instrument manufacturers in the development and improvement of instrumentation. He is co-author of 6 patents, and over 100 peer-reviewed publications. To contact the author please email him directly at: [email protected]
Larry T. Taylor is Emeritus Professor of Chemistry, Virginia Tech (Blacksburg, VA). He is a member of the editorial boards for the Journal of Chromatographic Science, Chromatographia, and the Journal of Supercritical Fluids. He was a member of the organizing committee for the 4th through the 10th International Symposia on SFC/SFE. More recently he has served as the Chair of the Scientific Committee for SFC 2008 (Zurich), SFC 2009 (Philadelphia), and SFC 2010 (Stockholm). Larry is the author or co-author of approximately 400 peer reviewed publications and 12 patents. He presently serves as co-teacher of short courses addressed to SFC and SFE. To contact the author, please email him directly at: [email protected]
Daniel R Brannegan is currently a Senior Scientist at Analytical Development in Pfizer Global R & D. He joined Pfizer as a lab scientist in 2000 and worked on numerous drug candidates in early stage development. Recently, he has worked as an analyst for formulation work, including product enhancements projects and progression of non solid dosage formulations through development. Daniel received his B.A in chemistry from the College of the Holy Cross, Worcester MA in 1996, and his M.S in analytical chemistry from Virginia Tech in 2000.
Jian Wang, Ph. D. is currently an Associate Research Fellow at Analytical Development in Pfizer Global R & D. He joined Pfizer as a lab supervisor in 1998 and worked on numerous drug candidates as the analytical team leader in drug substance and a variety of formulations – tablets/capsules, parenteral products and devices, and in different development phases (Preclinical – phase III). In last few years, he has been involved in polytides portfolio support in Pfizer, led the analytical team for oligonucleotide platform development. Jian has 15 years of pharmaceutical R&D experience. Prior to joining Pfizer, he had worked at Johnson and Johnson for over three years. Jian received his Ph.D. in analytical chemistry from Emory University.