Dissolution is the primary pharmaceutical test that is designed to probe the performance of dosage forms. From its beginnings in the middle of the twentieth century to address some serious issues with products on the market [1], the scope of application has grown to include use as a tool for formulation selection during product development and as a tool to enable waivers of bio-studies [2,3] under certain circumstances. Naturally, a dissolution test should be well suited to its intended purpose, but the purpose may be different depending on the circumstances. The purpose of this article is to provide some insight into the development of dissolution tests for pharmaceutical dosage forms for several of the most frequent intents.
In order to successfully develop a dissolution test, we should first identify the Analytical Target Profile [4]: purpose of the test, expectations for the data generated, and how it will be decided if the test is meeting its intended purpose. A typical ATP for a quality control dissolution test is shown in Figure 1.
 Figure 1. ATP for a QC dissolution test for an imediate release tablet
Figure 1. ATP for a QC dissolution test for an imediate release tabletSubsequent steps typically include learning about the solubility of the drug, selecting the dissolution test conditions and developing an analytical procedure to measure the dissolved drug in solution.
Dissolution tests may be developed for a number of reasons: as a QC test for an immediate release product, as a test designed to aid in formulation selection using biorelevant dissolution or as a group of tests designed to facilitate obtaining a biowaiver. These are probably the three most frequent uses of dissolution tests in the pharmaceutical industry, although there may be others as well. In an ideal world, a single dissolution test might be able to address all three of these needs, but this is probably not realistic from a practical perspective. There is a growing interest on the part of regulators to see ‘clinically relevant dissolution specifications’ [5], that is, specifications that can be linked to the performance of the drug product in vivo, but we have a long way to go in this arena.
Starting with the QC dissolution test, the first order of business will be to collect information. What is known about the dosage form? Is it intended to be an immediate release, extended release or delayed release oral product, or something different? This will help to identify the time scale for the dissolution test, and could influence other parameters of the test (e.g. delayed release products have a fairly specific dissolution protocol described in the United States Pharmacopeia). Solubility is a key parameter for dissolution, so solubility should be determined empirically at several pH values across the physiological range (pH 1 to 7.5) (Fig. 2). There are many ways to conduct this experiment; one useful method is to put excess drug into a 30 mL vial with compendial buffers over the pH range and store them at 37°C or room temperature for 24 hours, with gentle agitation such as a rocking shaker. Are there any significant solution stability issues? These may manifest themselves during the testing of the solution stability samples.
 Figure 2. Determine the solubility as a function of pH, and compare this to ‘sink’ conditions (solubility equal to three times the concentration of a fully dissolved tablet). If the tablet potency is 100 mg, and the volume is 900 mL, ‘sink’ conditions are 333 ug/mL. Both pH 6.5 and 7.5 would be acceptable, since solubility exceeds ‘sink’ conditions.
Figure 2. Determine the solubility as a function of pH, and compare this to ‘sink’ conditions (solubility equal to three times the concentration of a fully dissolved tablet). If the tablet potency is 100 mg, and the volume is 900 mL, ‘sink’ conditions are 333 ug/mL. Both pH 6.5 and 7.5 would be acceptable, since solubility exceeds ‘sink’ conditions.Armed with this information, you are ready to start the dissolution test development process. The ATP stipulates use of a standard compendial dissolution apparatus, and the compendial General Chapters can be a valuable source of information [6, 7]. The rationale for this is most likely to facilitate transfers among different laboratories, and to improve the probability of acceptance by a regulatory agency. Using this approach, an apparatus, a stirring speed, a dissolution medium and sampling time points must be selected. While there are 7 different apparatuses described in USP, almost all of the monograph tests for dissolution utilize Apparatus 1 (baskets) or Apparatus 2 (paddles), so it makes sense to start there. The preferred stirring speeds are 100 rpm for baskets and 50 rpm for paddles, making these good speeds for initial development experiments. The speed can be adjusted based on preliminary results (e.g. to adjust the rate of release or to avoid coning, a phenomenon where insoluble constituents of the dosage form create an inverted cone in the bottom center of the dissolution vessel). The medium is usually a simple solution that is similar to the fluids found in the gastrointestinal tract. Hence, buffers in the pH 1-7.5 range are good choices. (Note: it is a good idea to avoid pH values around a pKa of the drug, where a fraction of the drug is ionized and the remainder is un-ionized, since this can lead to reproducibility issues.) Utilize the solubility data to select an appropriate starting point. It is best to select a pH where the dissolution medium will be able to dissolve at least three times the amount of drug present in the vessel, since it is known that as a solution nears saturation, the rate of solubilization can slow down. ‘Sink conditions’, the ability to dissolve signifi cantly more drug than will be present in the dissolution vessel, allow the test to refl ect the properties of the drug product, rather than just refl ecting the intrinsic dissolution of the drug substance. If solubility in aqueous buff ers is insuffi cient, surfactants can be used to enhance solubility. Sodium lauryl sulfate and polysorbate 80 are used most frequently, although many other surfactants have also been used. When using surfactants, the minimum eff ective concentration should be used, and it may be necessary to also control the pH using an appropriate buff er.
For an immediate release product, typically samples are taken initially at 15 minute intervals (e.g. 15, 30, 45 and 60 minutes). A pragmatic approach is to perform a dissolution test using these time points, and evaluate the results. If the drug is fully dissolved (generally considered to be 85% or more) within 30 minutes, consider adding earlier time points such as 10 and 20 minutes (Fig. 3). Samples are taken at multiple time points, sometimes called ‘profi les’, during development with a goal of reducing this to one or two time points with specifi ed acceptance criteria during routine testing.
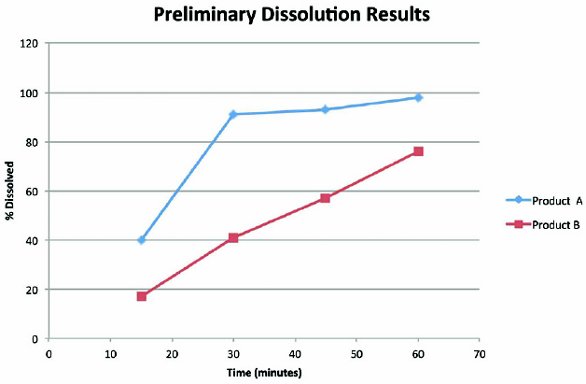 Figure 3. In these experiments with immediate release products, preliminary dissolution results showed the need to modify the sampling times. Product A dissolves within 30 minutes, so it would be appropriate to add 10 and 20 minute time points to fully characterize the profi le. Product B does not reach 85% dissolved within 60 minutes, so it would be appropriate to speed up the dissolution, either by increasing the agitation speed or selecting a pH where the solubility is higher.
Figure 3. In these experiments with immediate release products, preliminary dissolution results showed the need to modify the sampling times. Product A dissolves within 30 minutes, so it would be appropriate to add 10 and 20 minute time points to fully characterize the profi le. Product B does not reach 85% dissolved within 60 minutes, so it would be appropriate to speed up the dissolution, either by increasing the agitation speed or selecting a pH where the solubility is higher.If the product is intended for delayed release, dissolution is first carried out in Simulated Gastric Fluid to demonstrate that the drug will not be released into the stomach, and then switched to Simulated Intestinal Fluid (or other medium appropriate for the particular drug). More details on dissolution testing of delayedrelease drug products can be found in USP General Chapter <711>. Extended release products are designed to have a longer period of dissolution, so the test may be carried out for several hours or a full day or even longer (for instance, in the case of drugeluting stents). The goal for an extended-release dissolution test is generally to identify three time points: an early time, which is used to demonstrate that there is not ‘dose dumping’ or a rapid, unintended release of the drug, an intermediate time, which is used to demonstrate that the release of the drug is within a certain range of % dissolved values, and a later time point, which is used to demonstrate that the drug is fully released.

The analytical technique chosen to determine the amount of drug in the dissolution samples is usually selected to provide suitable accuracy and precision while minimizing time or other resources. Frequently spectrophotometric analysis, such as UVVis spectrophotometry, will be chosen if the drug has appropriate absorptivity and the formulation components do not interfere with the analysis. Another commonly selected technique is HPLC, which may have advantages of a larger range of concentrations which can be analyzed without dilution and easier data handling using electronic chromatographic data systems (CDS).
As with any analytical method, there may be subtle issues that can affect the acceptability of the results. In the case of dissolution testing, some of the common areas of concern include solution stability, the impact of dissolved air (which can be controlled by deaeration of the medium, which is discussed in USP General Chapter <711>), the timing and location of sample withdrawal and clarification of the samples (effectively removing any undissolved particles from samples which will be analyzed, since these can continue to dissolve and give erroneously high results).
An interesting aspect of dissolution testing is the unique, dynamic nature of the test procedure. This can make interpretation of the test results challenging. By way of contrast, consider the assay of a batch of tablets. In that case, there is a certain amount of drug in the tablets, and the job of the assay is to measure that with an acceptable degree of certainty, which can be quantitatively evaluated using accuracy and precision determinations. With dissolution testing, it is possible to design many different test conditions which may give widely varying results; how do you know which is the most appropriate set of conditions? This question actually leads us to the possibility of different goals for a dissolution test.
The most common goal is to develop a quality control test, which will provide a reasonable indication of whether or not the batch in question is similar in behavior to some reference batches, typically material used in pivotal clinical studies. A well-designed dissolution test will have been evaluated for its discriminating power: can it detect differences in manufacturing or physical properties, such as varying levels of lubricant or changes in tablet hardness? On the other hand, some dissolution tests are over-discriminating: seemingly insignificant differences between batches may result in large differences in dissolution results, especially if results from only a single time point are examined. These reasons make it very important to develop a good understanding about the ability of the test to detect changes while not providing misleading results due to over-discrimination.
Ideally, there would be a relationship between dissolution results and in vivo performance. Although this concept has been touted for decades, it can be difficult or impossible to develop to develop such a relationship, called an in vivo in vitro correlation, especially for immediate release products. More recently, the FDA has suggested exploring ‘clinically relevant specifications’, including one approach where various batches, representing the expected range of manufacturing variation and dissolution results, are evaluated in vivo. If the in vivo data suggest the batches within these ranges are acceptable, that can provide justification for wider dissolution specifications.
Another use for dissolution testing can be during formulation selection. During formulation development, how can the optimal formulation be selected from multiple iterations? One approach is to use dissolution conditions which closely mimic physiological conditions and compare the results among the various formulation iterations. Significant work has been done with ‘biorelevant media’ which may be useful in this situation [5,6].
One way in which in vitro dissolution has proved valuable to the industry is through its ability to obtain biowaivers. The ‘gold standard’ for demonstrating that two different formulations are equivalent is by demonstrating in vivo bioequivalence. In vivo testing is very time consuming and expensive, and the regulatory agencies have agreed that, in certain situations, if in vitro results are similar, it is possible to obtain a waiver of bioequivalence studies [7- 9]. Typical situations are where the changes to the formulation are minor and dissolution results are similar across the physiological range of pH values (Fig. 4).
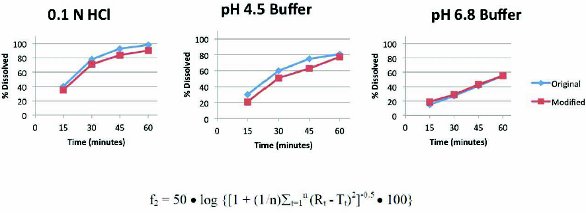 Figure 4. In this case, dissolution data were generated in three media (conditions were 50 rpm paddles) to support a request for a biowaiver. Since the results in each of the media were similar (i.e. f2 was greater than 50) and the drug was not a narrow therapeutic agent, it is likely the request would be successful.
Figure 4. In this case, dissolution data were generated in three media (conditions were 50 rpm paddles) to support a request for a biowaiver. Since the results in each of the media were similar (i.e. f2 was greater than 50) and the drug was not a narrow therapeutic agent, it is likely the request would be successful.Gaining insight into the performance of a drug product can be very valuable, and it is important to have a means of demonstrating that the performance is consistent from one batch of a drug product to another. Dissolution testing remains the best tool that we have to evaluate this, but it can be very challenging to develop a test which provides all of the necessary information. Good science is critical in choosing the test conditions, challenging their appropriateness and interpreting the results. Pharmaceutical scientists and regulators continue to rely on dissolution testing as a surrogate for performance, and the approaches for development and evaluation of these tests continue to improve and evolve. Dissolution testing is an expected test for Quality Control, but can also be very useful for formulation selection and to assist in waiver of bioequivalence testing when minor changes in formulation are considered.
Author Biography
Greg Martin is President of Complectors Consulting (www. complectors.com) which provides consulting and training in the area of Pharmaceutical Analytical Chemistry. He has over has over 25 years of experience in the pharmaceutical industry and volunteers for USP. He serves as Vice Chair of the General Chapters – Physical Analysis Expert Committee, and is on the Expert Panel for Use of Enzymes for Dissolution and Testing of Gelatin Capsules. He is also Chair of the AAPS In Vitro Release and Dissolution Testing Focus Group. He can be contacted at greg.martin@complectors.com.
References
- Dokoumetzidis, Aristides and Macheras, Panaos, A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System, International Journal of Pharmaceutics 321 (1 and 2), Sep 2006
- Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate Release Solid Oral Dosage Forms Based on a Biopharmaceutical Classification System. CDER, FDA, Aug. 2000.
- Guidance for Industry: Immediate Release Solid Oral Dosage Forms: Scale Up and Post Approval Changes. CDER, FDA, Nov. 1995.
- Nethercote, P., Borman, P., Bennett, T., Martin, G., and McGregor, P., “QbD for Better Method Validation and Transfer,” Pharmaceutical Manufacturing, 9(4), 2010.
- Boni, Julia Elisabeth, Brickl, Rolf Stefan, Dressman, Jennifer and Pfefferle, Martin L. Instant FaSSIF and FeSSIF—Biorelevance Meets Practicality. Dissolution Technologies, August 2009, p. 41-45.
- Fotaki, Nikoletta and Vertzoni, Maria, Biorelevant Dissolution Methods and Their Applications in In Vitro - In Vivo Correlations for Oral Formulations, The Open Drug Delivery Journal, 2010, 4, 2-13.
- Sharp, Sandra, “Setting Clinically Relevant Dissolution Specifications: A Regulatory Perspective”, presented at AAPS Annual Meeting, Chicago, IL, Oct 2012
- USP 35 – NF 30. General Chapter <711> Dissolution
- USP 35 – NF 30. General Chapter <1092> The Dissolution Procedure: Development and Validation