Advances in the field of Advanced Therapy Medicinal Products (ATMPs) have given rise to this area of patient disease control and cure response, Figure 1. These advances have the shape of cellular and molecular biotechnology progress that affects gene therapy, somatic cell therapy, and tissue engineering. ICH Q92 is the standard for Quality Risk Management (QRM), including ATMPs.
The United States FDA has issued; Chemistry, Manufacturing, and Control (CMC) information for Human Gene Therapy Investigational New Drug Applications (INDs).1 This guidance focuses on the following points:
- Administrative Information (Module 1 of the CTD)
- Summary of Quality Information (Module 2 of the CTD)
- Manufacturing Process and Control Information (Module 3 of the CTD)
In the European Union, the EMA has regulation on advanced therapy medicinal products (known as ATMPs Regulation)3
This ATMPs Regulation includes specific guidance for ATMPS:
- implementing a pharmacovigilance system
- risk identification
- risk minimization measures
- post-authorization Safety & Efficacy studies
- management and reporting of adverse reactions
- evaluation of the effectiveness of the risk management system
Managing the Risks of ATMPs
Advanced Therapy Medicinal Products (ATMPs) are evaluated using a risk-based approach. This approach considers various risks associated with the clinical use of ATMPs, including those related to quality, safety, and efficacy. The risks are determined by factors such as the ATMP’s purity, biological activity, and application.7
The clinical use of ATMPs in humans may be associated with specific risks to the patient and third parties. These risks are determined by various risk factors, which are related to the quality, biological activity, and application of the ATMPs. Since ATMPs are very diverse (i.e. gene therapy medicinal products (GTMPs), somatic cell therapy medicinal products (sCTMPs), tissue engineering products (TEPs), and combined ATMPs. A flexible approach to address and evaluate potential risks associated with the clinical use of ATMPs is described in the “Risk-Based” approach.
The concept of a “Risk-Based approach” has been introduced to the legislation with the revision of Annex 1, part IV of Directive 2001/83/ EC7 as amended by Directive 2009/120 EC.8 The aim of the risk-based approach in the development of ATMPs is to determine the extent of quality, non-clinical, and clinical data to be included in the Marketing Authorization Application (MAA), by the scientific guidelines relating to the quality, safety, and efficacy of medicinal products and to justify any deviation from the requirements of this Annex.
The application of the risk-based approach in the preparation of an MAA dossier is optional. However, in cases where the risk-based approach is being applied, the applicant is advised to follow the methodology as laid down in the present guideline.7
As with other medical products, risk detection should start as early as possible and continue throughout the development. This will allow us to minimize and prevent risks when it’s possible!
Thus, the safety and efficacy are managed through the risk management plan, Figure 2.
For ATMP,s the scope of the risks is a bit different than traditional pharma and biopharma, for example:
- Quality risks related to characteristics, storage, or distribution;
Disease transmission due to cell/tissue origin
Tumorigenicity – that is, the risk of generating tumors due to unwanted mutations
Problems in preservation, freeze,ng and thawing related to cold chain or other controlled temperature conditions
- Risks due to patient conditions, underlying disease, or parallel treatment;
Anaphylactic shock or graft rejection
Genetic modification of the patient’s cells
Infections caused by viral vectors used in gene therapy medicinal products
- Risks to patients due to administration;
Dosing errors
Repeated surgical or administration procedures
Use of medical devices
- Risks due to the persistence of the product in the patient;
Availability of rescue procedures, antidotes, and their risks
Late complications like malignancies and autoimmunity
Impact of Previous Therapies
- Risks to healthcare professionals or anyone in close contact with the patient;
Risks of dissemination and transmission to the environment
Risk identification is then followed by the construction of the Risk Management Plan, the benefit t-risk assessment identification, and then taking risk minimization measures, Figure 3. Then it is required to measure the effectiveness of these risk minimization measures.
Why Would You Need a Digital QRM System for ATMPs
For ATMP product(s) it is required to establish a quality Target Product Profile (QTPP). Then, from there, build a list of Critical Quality Attributes (CQA) and Critical Process Parameters (CPPs). As a result, by keeping a level of measurement and control over these CQAs and CPPs, you can make sure the ATMPs produced have the desired quality.
The question is: What is the most important aspect of the QRM process? It is believed that the key point is to have a standard application of risk management procedures. The reasons are simple: simplification, replicability, data and information integrity, and the possibility to use information through several processes.
The challenge here is having that standard application of risk management principles. Where having a Digital QRM System for ATMPs comesinton play.
If you have a digital QRM platform where all the information is centralized and shared, it can bring a whole lot of benefits;
- Significantly reduce the time needed to make the risk assessments
- Support a much faster documentation system
- Reduce the effort and resources necessary to manage the risks of your processes and products.
- Leverage already existing information as a foundation for new product development.s
Risk-Based Approach
ATMPs are complex products and risks may differ according to the type of product, nature/characteristics of the starting materials, and level of complexity of the manufacturing process, Figure 4. It is also acknowledged that the finished product may entail some degree of variability due to the use of biological materials and/or complex manipulation steps; e.g., cultivation of cells, manipulations that alter the function of the cells, etc. In addition, the manufacture and testing of autologous ATMPs (and allogeneic products in a donor-matched scenario) poses specific challenges, and the strategies implemented to ensure a high level of quality must be tailored to the constraints of the manufacturing process, limited batch sizes, and the inherent variability of the starting material.4
ATMPs are at the forefront of scientific innovation and the field is experiencing rapid technological change that also impacts the manufacturing processes. For instance, new manufacturing models are emerging to address the specific challenges of ATMPs, e.g.; decentralized manufacturing for autologous products). Additionally, ATMPs are also often developed in an academic or hospital setting operating under quality systems different from those typically required for the manufacture of conventional medicinal products, Figure 4.
It follows that, in laying down the GMP requirements applicable to ATMPs, it is necessary to recognize a certain level of flexibility so that the ATMP manufacturer can implement the measures that are most appropriate having regard to specific characteristics of the manufacturing process and of the product. This is particularly in Figure 4. Overview of a Typical Quality Risk Management Process is important in the case of investigational ATMPs, especially in early phases of clinical trials (phase I and phase I/II), due to the often incomplete knowledge about the product (e.g., potency) as well as the evolving nature of the routines to adjust the manufacturing process to the increased knowledge of the product.
Application of the Risk-Based Approach by ATMP Manufacturers
The risk-based approach (“RBA”) applies to all types of ATMPS. The quality, safety, and efficacy attributes of the ATMPs and compliance with GMP should be ensured for all ATMPs, regardless of whether they are developed in a hospital, academic, or industrial setting, Figure 5.4
Manufacturers are responsible for the quality of the ATMPs they produce. The risk-based approach permits the manufacturer to design the organizational, technical, and structural measures that are put in place to comply with GMP and thus to ensure quality according to the specific risks of the product and the manufacturing process. While the risk-based approach brings flexibility, it also implies that the manufacturer is responsible for putting in place the control/mitigation measures that are necessary to address the specific risks of the product and the manufacturing process.
The quality risks associated with an ATMP are highly dependent on the biological characteristics and origin of the cells/tissues, the biological characteristics of the vectors; e.g., replication competence or reverse transcription and transgenes, the level and characteristics of the expressed protein, for gene therapy products, the properties of other non-cellular components (raw materials, matrices), and the manufacturing process.
When identifying the control/mitigation measures that are most appropriate in each case, the ATMP manufacturer should consider all the potential risks related to the product or the manufacturing process based on all information available, including an assessment of the potential implications for the quality, safety, and efficacy profile of the product, as well as other related risks to human health or the environment. When new information emerges that may affect the risks, an assessment should be made whether the control strategy; i.e. the totality of the control and mitigation measures applied, continues to be adequate.
The evaluation of the risks and the effectiveness of the control/ mitigation measures should be based on current scientific knowledge and accumulated experience. This risk evaluation is linked to the protection of patients.
The application of a risk-based approach can facilitate compliance but does not obviate the manufacturer’s obligation to comply with relevant regulatory requirements and to demonstrate that it can adequately manage the risks of the product/manufacturing process. It likewise does not replace appropriate communications with the authorities.4
Risk-Based Approach in Connection with Raw Materials
The application of the risk-based approach is used when determining the strategy to ensure the quality of the raw materials.4 The application of the risk-based approach requires that the manufacturer has a good understanding of the role of the raw material in the manufacturing process and, in particular, of the properties of the raw materials that are key to the manufacturing process and the final quality of the product.
It is important to consider the level of risk of the raw material due to the intrinsic properties thereof; e.g. growth factors, basic media, culture media containing cytokines, basal media without cytokines, raw material from animal origin, autologous plasma, or the use thereof in the manufacturing process, higher risk if the raw material comes into contact with the starting materials.
Finally, it needs to be assessed if the control strategy; e.g., qualification of suppliers, and the performance of suitable functional testing is sufficient to eliminate the risks or to mitigate them to an acceptable level.
Starting Materials
According to the European Union, the donation, procurement, and testing of human tissues and cells used as starting materials should be by Directive 2004/23/EC.11 For blood-derived cells, compliance with Directive 2002/98/EC12 regarding donation, procurement, and testing is likewise acceptable. The accreditation, designation, authorization, or licensing of the supplier of starting materials as provided for under the legislation above-referred should be verified. When the cells/tissues used are outside the scope of Directive 2004/23/EC or as appropriate Directive 2002/98/EC; e.g. cell lines/cell banks established outside the EU, or cells procured before the entry into force thereof, the ATMP manufacturer or as appropriate, the sponsor or marketing authorization holder, should take appropriate steps to ensure the quality, safety and traceability thereof, by the terms of the marketing authorization/ clinical trial authorization.
The ATMP manufacturer (or, as appropriate, the sponsor or marketing authorization holder) should establish quality requirements for the starting materials (specifications) which should be agreed upon with the supplier(s). These agreed specifications should cover aspects of the production, testing and control, storage, and other aspects of handling and distribution as appropriate. Depending on the product’s characteristics, testing in addition to that foreseen in Directive 2004/23/EC or as appropriate Directive 2002/98/EC may be required. The agreed specifications should comply with the terms of the marketing authorization or clinical trial authorization. The ATMP manufacturer should verify compliance with the supplier’s materials Figure 5. Overview of a Typical Quality Risk Management Process with the agreed specifications. The level of supervision and further testing by the ATMP manufacturer should be proportionate to the risks posed by the individual materials. Blood establishments and tissue establishments authorized and supervised by Directive 2002/98/EC or Directive 2004/23/EC do not require additional audits by the ATMP manufacturer regarding compliance with the requirements on donation, procurement, and testing provided for under the national law of the Member State where the blood/tissue establishment is located. However, if the agreed specifications foresee additional requirements; e.g. additional testing, adequate supervision concerning the additional requirements should be carried out. In addition to the specifications for the starting materials, the agreement between the ATMP manufacturer or as appropriate, the sponsor or marketing authorization holder and the supplier including blood and tissue establishments should contain clear provisions about the transfer of the information regarding the starting materials, in particular, on tests results performed by the supplier, traceability data, and transmission of health donor information that may become available after the supply of the starting material and which may have an impact on the quality or safety of the ATMPs manufactured therefrom.
The risk of contamination of the starting materials during their passage along the supply chain must be assessed, with particular emphasis on viral and microbial safety and Transmissible Spongiform Encephalopathy (“TSE”). Compliance with the latest version of the Note for Guidance on Minimizing the Risk of Transmitting Animal Spongiform Encephalopathy (TSE) Agents via Human and Veterinary Medicinal Products is required. Only starting materials that have been released by the person responsible for quality control should be used. Where the results from the test(s) required to release the starting materials take a long time; e.g., sterility test, it may be permissible to process the starting materials before the results of the test(s) are available. The risk of using a potentially failed material and its potential impact on other batches should be assessed and understood. In such cases, the finished product should only be released if the results of these tests are satisfactory unless appropriate risk mitigation measures are implemented. Starting materials in the storage area should be appropriately labeled. Labels should bear at least the following information:
- the designated name of the product and the internal code reference (if applicable)
- a batch number is given on the receipt
- storage conditions
- the status of the contents; e.g., in quarantine, on test, released, rejected
- an expiry date or a date beyond which retesting is necessary
When fully computerized storage systems are used, all the above information need not necessarily be in a legible form on the label. The use of automated systems; e.g., the use of barcodes, is permissible.
Risk-Based Approach in Connection With the Testing Strategy
It is acknowledged that in some cases it may not be possible to perform the release tests on the active substance or the finished product, for example, due to technical reasons; e.g., it may not be possible to perform the release tests on the combined components of certain combined products, time restrictions; i.e., the product needs to be administered immediately after completion of manufacturing, or when the amount of available product is limited to the clinical dose.4
In these cases, an adequate control strategy should be designed. For example, consideration can be given to the following options;
- Testing of key intermediates (instead of the finished product) or in-process controls (instead of batch release testing) if the relevance of the results from these tests to the critical quality attributes of the finished product can be demonstrated.
- Real-time testing in case of short shelf-life materials/products.
- Increased reliance on process validation. When the scarcity of materials or the very short shelf-life limits the possibilities for release controls, the limitations should be compensated by a reinforced process validation (e.g., additional assays, such as potency testing or proliferation assays may be performed after batch release as supporting data for process validation). This may also be relevant for investigational ATMPs. While process validation is not expected for investigational medicinal products, it may be important when routine in-process or release testing is limited or not possible.
It is stressed that the release testing strategy should be performed by the marketing/clinical trial authorization.
The following examples may also be considered: The application of the sterility test to the finished product by the European Pharmacopoeia (Ph. Eur. 2.6.1) may not always be possible due to the scarcity of materials available, or it may not be possible to wait for the final result of the test before the product is released due to short shelf life or medical need. In these cases, the strategy regarding sterility assurance has to be adapted. For example, the use of alternative methods for preliminary results, combined with sterility testing of media or intermediate product at subsequent (relevant) time points could be considered.
The use of validated alternative rapid microbiological methods may also be considered. For example, sole reliance on alternative microbiological methods according to Ph. Eur. 2.6.27 may be acceptable when this is justified having regard to the specific characteristics of the product and the related risks, and provided that the suitability of the method for the specific product has been demonstrated.
If the results of the sterility test of the product are not available at release, appropriate mitigation measures should be implemented, including informing the treating physician.
- As cells in suspension are not clear solutions, it is acceptable to replace the particulate matter test with an appearance test (e.g. color), provided that alternative measures are put in place, such as controls of particles from materials (e.g. filtration of raw material solutions) and equipment used during manufacturing, or the verification of the ability of the manufacturing process to produce low particle products with simulated samples (without cells).
- It may be justified to waive the ongoing stability program for products with shorter shelf-life.
Risk Factors
A risk factor is defined as a “qualitative or quantitative characteristic that contributes to a specific risk following handling and/or administration of an ATMP.” Aspects that should be considered when identifying risk factors include, but are not limited to: the nature of the product, non-cellular components, biodistribution, manufacturing issues, and clinical aspects.
Examples of risk factors that can be associated with cell-based medicinal products could include, but may not be limited to: the origin of cells or tissues (autologous vs. allogeneic), the ability of cells to proliferate and differentiate, the ability to initiate an immune response (as target or effector), the level of cell manipulation (in vitro/ex vivo expansion/activation, genetic manipulation), aspects of the manufacturing process, non-cellular components, the mode of administration (ex vivo perfusion, local, systemic) and the duration of exposure (short to permanent).
Risk factors that can be associated with GTMPs depend on the vector as well as on the transgene expression cassette used, and, in the case of a cell-based GTMP also on the cell population to be genetically modified. Typical risk factors include, but may not be limited to: the potential of the vector for and its extent of chromosomal integration, vector immunogenicity, the capacity of the vector for latency/ reactivation and/or mobilization, and its potential for recombination/ re-assortment and biodistribution to non-target sites. Risk factors may also be attributable to the expression of the therapeutic or any other transgene delivered and to the duration of expression. In the case of a cell-based GTMP, risk factors as described for cell-based medicinal products may also be applicable. The replication-incompetence or competence of a vector and its capacity to inadvertently replicate after complementation by a respective wild-type or helper virus may also have to be taken into consideration as risk factors.
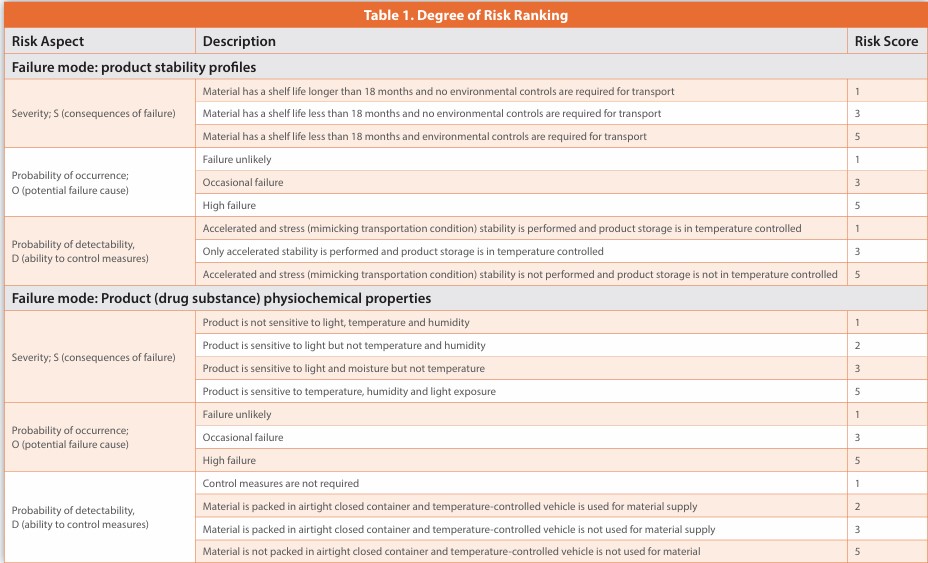
The clinical use of the ATMP should also be considered when identifying risk factors. Patient, disease, and medical procedure-related risk factors, including those associated with the administration of the ATMP, may contribute to a specific risk, Table 1.
Quick Response Manufacturing
Quick Response Manufacturing is a strategy for reducing lead times across all functions of an organization. The resulting improvements in speed and responsiveness increase the organization’s agility and responsiveness, resulting in a competitive advantage. Many well-known Lean Manufacturing tools have been developed for high-volume/low-variety, or ‘mass production’ environments. Think of techniques such as Pull Systems, Kanban, Line Balancing, and Heijunka for instance, often applied to fast-moving production lines. However, these tools often do not translate well to low volume/high variety environments, which require short batch runs, higher levels of customization, and fast response to changes in customer demand.
For businesses facing the challenge of meeting increased customization and speed, Quick Response Manufacturing is a strategy that relentlessly focuses on reducing lead time both on the shop floor and in the office operations.
The advent of immunotherapy, notably immune checkpoint blockers (ICB), has heralded a new era in cancer treatment, bolstering the body’s own defenses against tumors. The efficacy of these treatments hinges on the presence of potent T cells within the tumor microenvironment.
Categorizing tumors into distinct immune profiles reveals the challenges posed by “cold” tumors, which lack the critical T cell presence needed for immunotherapy success. In contrast, with its innate ability to reprogram immune cells, the innate immune pathway emerges as a promising frontier in overcoming tumor resistance. Targeted drugs for this pathway have faced hurdles in clinical translation.
To unlock the full potential of innate immune pathways, understanding their adaptability and diversity is crucial, guiding the way towards personalized precision medicine strategies for cancer treatment In the figure: Tumor immune microenvironment. The TME induces immune cells’ pro-tumor differentiation or polarization through various pathways. The abnormal microenvironment of a tumor, along with factors secreted by the tumor itself, can induce cells within this environment to undergo differentiation or polarization towards a pro-tumor phenotype, or experience exhaustion of their anti-tumor functions. However, some cytokines have the potential to overcome or reverse these pro-tumor phenotypes.
Quality Risk Management and Assurance of Potency
At all stages of the product lifecycle, you should use quality risk management to assess risks to product potency and to reduce those risks to acceptable levels. It is recommended to consider the following concepts when designing a potency assurance strategy for the product:
Quality target product profile (QTPP) - A QTPP should include a summary of the potency-related characteristics of the product. The QTPP should be developed based on an understanding of the product’s mechanism of action (MOA), the intended clinical indication, and the route of administration.
Control strategy - Manufacturing process controls and product quality controls play vital roles in potency assurance. These controls include process parameters, in-process testing, material testing or examination, lot release tests, and associated acceptance criteria.
Critical quality attribute (CQA) - Potency-related CQAs are attributes of the product that are important for achieving the intended therapeutic effect. You should identify the potency-related CQAs of the product to the extent needed to establish a phase-appropriate control strategy. Your manufacturing process should consistently produce lots that have all CQAs within appropriate pre-determined limits.
Critical process parameter (CPP) - CPPs are manufacturing process parameters that can influence CQAs. You should identify CPPs that may affect potency-related CQAs, and these CPPs should be monitored or controlled within appropriate pre-determined limits.
Risk assessment - You should identify risks to potency-related CQAs, analyze the probability and severity of these risks, and evaluate their significance. You should assess risks not only when initially designing the manufacturing process and control strategy, but also throughout the product lifecycle.
Risk reduction - Any risks to potency-related CQAs that are unacceptably high should either be avoided or reduced to acceptable levels by appropriately designing the manufacturing process and control strategy.
Potency Risk Assessment
As part of quality risk management, you should use formal risk assessment tools to assess risks to potency comprehensively. Risk assessments should start with identifying what might go wrong: what are the factors that might adversely affect the potency of the product both during and after manufacturing? The process of identifying risks to potency will be most effective when the product’s MOA and potency-related CQAs are well understood and the risks to potency-related CQAs can be determined with confidence. The risk assessment should include not only factors that may affect potency at the time of lot release, but also factors that may affect potency after lot release, such as the container closure, delivery devices, conditions for drug storage, shipping or handling, and conditions for thawing or preparing the drug for administration. Analyzing and evaluating risks to potency can be challenging if assays used to measure potency-related CQAs have not been qualified to determine whether they have adequate performance. Using unqualified assays may decrease the ability to analyze risks to potency, due to a potential for inconsistent assay performance or uncertainty about the ability of the assay to detect clinically relevant changes in product potency. Risks to potency should be reassessed as you increase your understanding of your product and manufacturing process. Before implementing manufacturing changes (including changes to materials), you should assess the risks that the changes may pose to product potency. Following the evaluation of risks, any risks to potency that are unacceptably high should be mitigated or avoided through the design of the manufacturing process and the control strategy.
Continued process verification. During manufacturing of a licensed product, you should routinely collect and analyze product and process data to verify that the manufacturing process remains in a state of control that assures potency. These analyses may suggest potential opportunities to improve potency assurance through adjustments to the manufacturing process or control strategy. In certain cases, potency assurance may also be improved by including additional testing as part of continued process verification. For products that have an extremely short shelf life with insufficient time to complete a potency bioassay before lot release, it should be possible to perform lot release testing for potency using physicochemical assays. It is recommended to initiate one or more potency bioassays immediately after manufacturing the drug product and evaluate the results when they become available post-release. For both investigational and licensed products, such post-release testing will help to verify that the manufacturing process is continuously capable of producing potent lots. The appropriateness and frequency of such post-release testing should be based on a risk assessment.
If one aspect of the potency assurance strategy cannot adequately mitigate a risk to product potency, then it should mitigate the risk by strengthening other aspects of the potency assurance strategy. For example, lot release testing may not be able to fully confirm potency if a product’s potency-related CQAs are poorly understood or difficult to quantitate, or if a product has an extremely short shelf life that does not allow enough time to perform a bioassay. In these cases, other aspects of the potency assurance strategy (such as process design and process control) will take on increased importance and should therefore be more stringent and extensive.5
Potency assurance for cellular and gene therapy products involves a risk-based strategy to ensure that these products are safe, efficacious, and capable of achieving the desired therapeutic effects in patients.1 The strategy includes:
- Manufacturing process design
- Manufacturing process control
- Material Control
- In-process testing
- Potency lot release assays
Prevention of Cross-Contamination in Production
An evidence-based QRM process should be used to assess and control the cross-contamination risks presented by the products manufactured. Factors to consider include:
- vectors used and the risk of occurrence of replication-competent virus (including different levels of risk derived from the use of replication-limited, replication-defective, conditional replication, and replication-incompetent vectors)
- facility/equipment design and use
- personnel and material flow
- microbiological and other adventitious agent controls
- characteristics of the starting materials/active substance and raw materials
- process characteristics
- clean room conditions
- cleaning processes
- analytical capabilities relative to the relevant limits established from the evaluation of the products
The outcome of the QRM process should be the basis for determining the process workflow and necessity for and extent to which premises and equipment should be dedicated or single-use systems should be used for a particular product. This may include dedicating specific product contact parts or dedication of the entire manufacturing facility. It may be acceptable to confine manufacturing activities to a segregated, self-contained production area within a multiproduct facility, was justified. Results should be reviewed jointly with the Contamination Control Strategy.9
References
- FDA Guidance Document, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), Guidance for Industry, January 2020; Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs)
- ICH guideline Q9 (R1) on quality risk management Step 5, EMA/CHMP/ICH/24235/2006 Committee for Human Medicinal Products
- Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004 (Text with EEA relevance); Regulation - 1394/2007 - EN - EUR-Lex (Europa.eu)
- EudraLex The Rules Governing Medicinal Products in the European Union Volume 4 Good Manufacturing Practice Guidelines on Good Manufacturing Practice specific to Advanced Therapy Medicinal Products; ad33d9dd-03f0-4bef-af53-21308ce2187d_en (Europa.eu)
- Potency Assurance for Cellular and Gene Therapy Products, Draft Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Biologics Evaluation and Research December 2023; SOPP 8002 Appendix 2 - Draft Guidance.doc (fda.gov)
- Guidance for Industry: Process Validation: General Principles and Practices; January 2011, Revision 1. https://www.fda.gov/media/71021/download
- Guideline on the risk-based approach according to annex I, part IV of Directive 2001/83/ EC applied to Advanced therapy medicinal products, 11 February 2013 EMA/CAT/ CPWP/686637/2011 Committee for Advanced Therapies (CAT); Guideline on the risk-based approach according to Annex I, part IV of Directive 2001/83/EC applied to Advanced Therapy Medicinal Products (Europa.eu)
- EUROPEAN PHARMACOPOEIA 6.0 2.6.1. Sterility; Print Preview - C:\DOCUME~1\xpress\ LOCALS~1\Temp\.aptcache\00b-prelim-roman-E/tfa03632 (uspbpep.com)
- Annex 2A, Guide to Good Manufacturing Practice for Medical Products Annexes, Pharmaceutical Inspection Convention, Pharmaceutical Inspection Co-operation Scheme PE 009-17 (Annexes) 25 August 2023 PE 009-17 (Annexes), 25 August 2023; PICS GMP Guide (Annexes) (picscheme.org)
- New Ph. Eur. General Charter Adopted: Raw materials of biological origin for the production of cell-based and gene therapy medicinal products, New Ph. Eur. General Charter Adopted: Raw materials of biological origin for the production of cell-based and gene therapy medicinal products - European Directorate for the Quality of Medicines & HealthCare (edqm. eu) Directive 2004/23/EC of the European Parliament and of the Council of 31 March 2004 on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells;
- Directive - 2004/23 - EN - EU Tissue Directive - EUR-Lex Europaa.eu)
- Directive 2002/98/EC of the European Parliament and the Council of 27 January 2003 setting standards of quality and safety for the collection, testing, processing, storage, and distribution of human blood and blood components and amending Directive 2001/83/EC; Directive - 2002/98 - EN - EUR-Lex Europaa.eu)
Author Details
Robert Dream- HDR COMPANY LLC
Publication Details
This article appeared in American Pharmaceutical Review: Vol. 27, No. 5July/Aug 2024Pages: 20-28
Subscribe to our e-newsletters
Stay up to date with the latest news, articles, and events. Plus, get special
offers from American Pharmaceutical Review delivered to your inbox!
Sign up now!