Background
Of all of the tests that we perform on pharmaceutical dosage forms, perhaps the most important, certainly the most physically taxing, is the dissolution test. Increasingly, the health authorities are asking more and more questions on submitted New Drug Applications (“NDAs”). Some of the questions that are being asked include; Is the dissolution method discriminating, and if so, what does it discriminate? Is the dissolution method bio-relevant? With these questions, and the potential for a significant amount of work that could go along with preparing the data for these experiments, increased efficiency in the laboratory is a necessity, if not a requirement. It is important to free the dissolution scientist of the mundane aspects of running the dissolution, in order for them to spend time designing the experiments and analyzing the data that they generate. Moreover, these scientists can perform other value-added activities such as method development and optimization.
The Dissolution Test
It is important to understand the basics of dissolution to discuss dissolution automation. The Webster’s dictionary defines ‘dissolution’ as, ‘the process by which a solid, liquid, or a gas is dispersed homogeneously into a gas, solid, or especially, a liquid.’ Over the course of history, the dissolution test has become more and more valuable in the determination of a pharmaceutical product’s clinical viability. The value of dissolution, or more importantly, the rate of dissolution, is used primarily as a formulation screening tool. Formulators use the dissolution test to distinguish between different variants of a dosage form. It is also used to determine product stability and serves as a quality control test to provide evidence of the physical characteristics of the product. Obtaining the required data for the formulation screening can lead to many dissolution tests, in different media and over long periods of time. A single formulation scientist can prepare enough formulations to keep two analytical colleagues busy for weeks testing the dissolution profiles. An efficient scientist in the dissolution laboratory can perform a maximum of four to six manual dissolution tests per day. A manual dissolution test is a labor-intensive process that requires many activities. The chemist is required to prepare the medium necessary for the testing, which includes solublizing the necessary ingredients, checking and adjusting the pH, de-aerating the media, pouring the media into the vessels, waiting for the media to warm, verifying the temperature is within the requirements (37.0 ± 0.5°C), dropping the dosage form, sampling an aliquot at specified times, and preparing an HPLC for injection of samples. Dissolution run times can vary from as short as 30 minutes to as long as several hours. Other mundane duties include cleaning the dissolution tester, washing and drying the glassware, and insuring cleanliness so that there is no carryover to the next experiment. All of these activities lend themselves nicely to being automated. These scientists’ time can be much better utilized, perhaps working on data analysis, preparing answers to health authority questions, working on development for the next compound, or validating the current testing methods. Alternatively, the dissolution test can be performed either fully automated or semi-automated. Fully automated dissolution tests can free the dissolution scientist from about ninety percent of the mundane exercises in the laboratory.
Automation
First, the question needs to be asked: “Why even automate the dissolution process?” As a laboratory scientist, we are being asked to do more in less time. The manual dissolution process is about as lean and efficient as it’s going to get. Formulation scientists need the data faster to get the right formulation so that the first clinical trial dates can be met, which are being started sooner and sooner. The pressure on the pharmaceutical industry to provide drugs at cheaper prices is ever increasing. Automating the dissolution test, while initially expensive, based on the cost of the up-front investment, reduces the cost per dissolution in the long run. The robot can perform the dissolution while the chemist is completely out of the lab. The dissolution test is highly regulated; automating the process provides a more consistent manner of performing the test.
There are several types of fully and semi-automated dissolution systems on the market that can be used in the dissolution laboratory today. Fully automated systems are most efficient when used for immediate release dosage forms while semi-automated systems provide the ability to sample from extremely long dissolution tests (i.e., Extended or Modified Release products).
The fully automated system consists of a dissolution tester, automated dissolution component, a robotic arm capable of attaching and removing USP Apparatus I baskets, a basket turret, a media switching valve (capable of delivering 4 types of different media to the dissolution bath), an autosampler, a sample collection device, a column switcher (if necessary) and an HPLC instrument. The column switching device is not usually necessary, as a suitable HPLC method or Flow Injection method would be developed before the dissolution testing progresses. This fully automated setup can run a maximum of 8 single point dissolutions or up to four multi-point profile dissolutions per evening. The steps that the dissolution chemist needs to perform on a daily basis are reduced drastically by using this system. While they are still charged with the preparation of dissolution medium, the dosage form insertion (basket addition or tablet dropping), sampling, filtering, and clean-up are left to the system.
Table 1
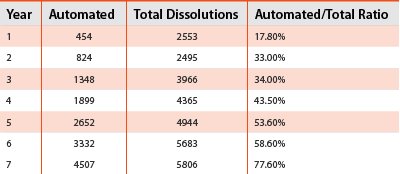
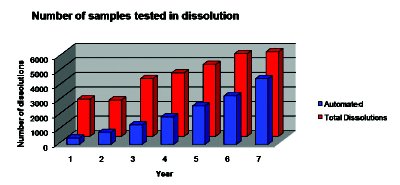
Figure 1
While the requests for dissolution data have increased dramatically in recent years, in an effort to control costs in the industry, the resources needed to perform these tests and obtain the data have remained constant. The automated systems have been able to keep up with the increased demand for dissolution data. A multi-year analysis of the number of dissolution tests performed at Novartis Pharmaceuticals Corporation are described and illustrated in the document. The data show that over the course of seven years the total number of dissolution tests in the laboratory increased by more than double from just over 2500 to more than 5800 dissolution tests. However, the lion’s share of the dissolution testing was performed by the automated system as shown in Table 1, and the chart in Figure 1. The Automated/Total dissolution ratio shows that greater than 75% of the dissolution tests in this laboratory were being performed by the automated systems in year 7.
Along with the increase in number of dissolution tests that were performed over the course of the years, the quality of the dissolution data has risen as well. The automated system reduces analyst-to-analyst variability, as well as the issues that can arise from different sampling positions, times, and tablet drops. United States Pharmacopeia, USP <711>, states that the volume of the dissolution medium to be placed within each vessel must be within ±1% of the stated volume in the test method. Manually, this is done using either a Class A graduated cylinder or a “to-dispense” volumetric flask. These different types of glassware have their own tolerances which add to the error of the manual dissolution test. The automated dissolution system dispenses the medium gravimetrically, which introduces less error and is done consistently from vessel to vessel and run to run. This is also important when replacing media that has been removed from sampling. A dissolution chemist might use a disposable syringe with graduations, however not calibrated, whereas the automated system would do this gravimetrically as well. Additionally, automation uses the same equipment for every run which eliminates the error which could be found from using different glassware. More importantly, USP <711> also states that the sample is to be taken midway between the surface of the dissolution liquid and the top of the spinning paddle blade or basket and not less than 1cm from the side of the vessel wall. This is where automating the dissolution test is most favorable. The automated systems available to the industry are calibrated to sample from the dissolution vessel in the same place every time. The manual procedure lacks this consistency. While the USP chapter designates a zone for sampling, reducing variability in the sampling process is most favorable.
Stability testing and QC type analysis comprise the bulk of the type of testing performed on these automated systems. However, automated dissolution systems are powerful method development tools for dissolution. Equipped with a media switching valve, the system can perform a variety of different experiments to help the analyst develop a robust method for any pharmaceutical product. These attachments to the system allow the user to connect up to 4 different media to the system and perform dissolution on the various products overnight. This same testing, done manually, would occupy the majority of 2-3 days depending on the chemist.

Figure 2
Like any analytical device, there is the possibility for errors, no system is without flaws. With the automated dissolution test however, the possibility exists that the errors would not be detected until after the HPLC analysis is complete. So the next question to be asked is, “How do we maintain and/or troubleshoot these devices?” Really, the answer is simple. First, we look for key performance indicators, such as successful overnight runs, extended testing without any errors, and we sporadically monitor the system during the day, so in short we must keep an “eye” on them. It is certainly not efficient to have an analyst monitor the system’s performance 24/7, the productivity gains would be instantly lost, but there is a way to monitor every movement of the system. A surveillance system is implemented to ensure that the system is working the way it is advertised. The system also monitors for random events such as basket drops or “double tablet” dropping into a single vessel. See Figure 2 for an image of the surveillance system. The surveillance system can provide answers to a significant number of laboratory investigations for dissolution values of “180%” or “0%”. Under any circumstance, values like these can call into question the formulation, and if the product is on stability, concerns about the product’s physical stability can arise. However, if after “going to the videotape,” can prove that there was a system malfunction, then the investigation can be closed quickly.
Overall, the surveillance system proves worth its weight in gold. An investigation into the number of successful dissolution runs compared to the number of “adverse events” (unsuccessful basket loading) over a period of nine months shows that the number of successful runs far outweighs the number of unsuccessful runs. The data are presented in Figure 3.
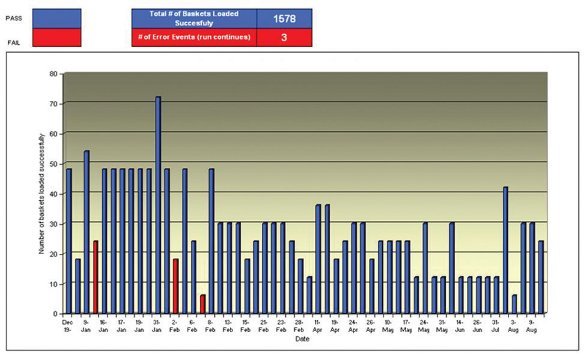
Figure 3
Finances
Financially, the initial investment of an automated dissolution system can be very high. The cost can range from $250,000 to $300,000 dollars for just the basic instrument. Including add-ons such as media switching valves and an on-line HPLC injection apparatus can drastically increase the price. The issue of cost however is greatly reduced when compared to that of dedicating a scientist or group of scientists to perform the duties required of repetitive dissolution. A simple look at the fact that the robot can perform 8 dissolution runs over one night whereas a highly efficient dissolution chemist can perform 4-5 dissolution runs in one day is evident that the cost per dissolution is reduced roughly by half.
Thinking “Out of the Box”
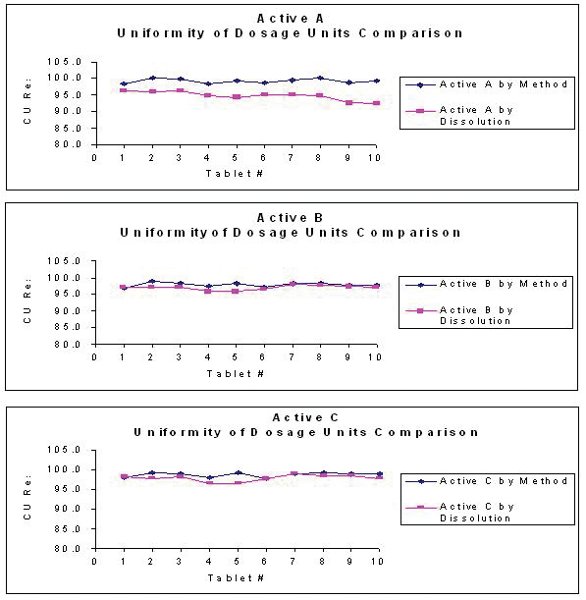
Figure 4
Table 2
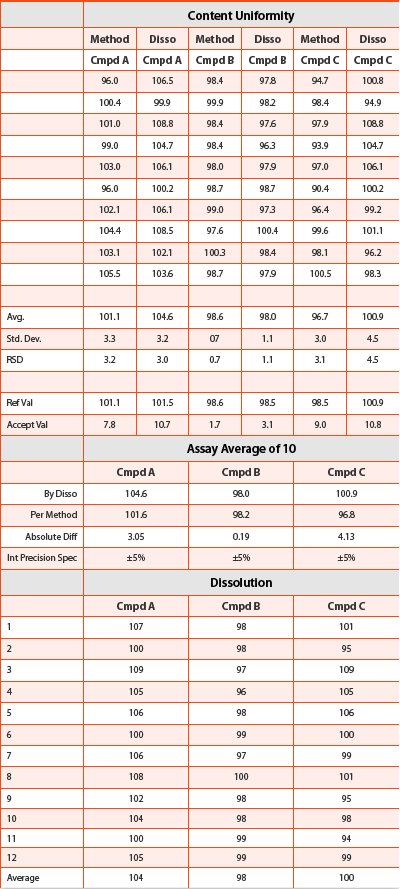
Automating the dissolution process clearly increases the efficiency of the laboratory. But, as scientists, we try to push the limits of what we can do with the instrumentation we have. Can we use the automated dissolution data to possibly provide data for such tests as Assay and Uniformity of Dosage? In our lab, we investigated the possibility of using the dissolution data generated by our automated systems to provide this information. If we test an n=12 dissolution and use the data from each of the first ten tablets for the data for Uniformity of Dosage, how does it compare to the data generated from the validated and compendial method for this test? Also, can we provide data for composite assay from the same set of experiments? The answer is, yes, it is possible. If we average the first ten tablets and compare that value with the value obtained from the validated assay method, what is the difference? Does it meet the criteria of Intermediate Precision? We investigated these questions using a triple active product and the data are provided in Figure 4 and Table 2.
The data are evaluated using internal SOP requirements for intermediate precision and USP<905> Uniformity of Dosage. The data analysis shows that the dissolution test pass the criteria listed for these documents, however, these are preliminary examinations of using dissolution data for evaluation of Assay and Uniformity of Dosage and further development and validation need to be investigated before using the data for GMP analysis.
Conclusions
It is clear that automating the dissolution laboratory dramatically increases productivity from both the standpoint of amount of samples being analyzed as well as the ability of the chemist to move on to other project-related activities. While we know that we can successfully automate these processes, it is important to observe and monitor the systems to keep them running flawlessly so that the integrity of the data produced does not come into question and this is easily accomplished by a simple surveillance system. It is also possible to increase efficiency by applying the automated dissolution test to other required testing for pharmaceutical dosage forms, however, more investigation is necessary to further validate this work.
References
- USP<711> Dissolution.
- USP<905> Uniformity of Dosage Units.
- Iarriccio, A. Automating the Dissolution laboratory. Presented at the Eastern Analytical Symposium, November 2010, Somerset, NJ.
- Iarriccio, A.; Kassis, A.; Paul, R.; Patel, T. Implementation, Qualification, and Validation of a fully automatic dissolution apparatus. Presented November 2007, at the Eastern Analytical Symposium, Somerset, NJ.
- Kassis, A.; Iarriccio, A.; Paul, R.; Bynum, K.; Patel, T. Increasing innovation and efficiency through automation in the pharmaceutical industry – scientific vs. business aspects. Presented November 2007 at the Eastern Analytical Symposium, Somerset, N.
Author Biography
Anthony Iarriccio is a Senior Scientist at Novartis Pharmaceuticals Corporation with over 12 years of pharmaceutical development experience. Specializing in HPLC analysis of pharmaceutical compounds, Anthony has expertise in developing and validating automated dissolution methods for many different compounds. He has an MS in Chemistry from the Polytechnic Institute of New York University and is currently pursing a Ph.D. in Materials Science from the same.
This article was printed in the May/June 2011 issue of American Pharmaceutical Review - Volume 14, Issue 4. Copyright rests with the publisher. For more information about American Pharmaceutical Review and to read similar articles, visit www.americanpharmaceuticalreview.com and subscribe for free.